Single-cell sequencing: an introduction to decoding cellular diversity
Written by Éva Mészáros
15. September 2025
Single-cell sequencing enables the analysis of molecular material at the level of individual cells. Unlike traditional bulk sequencing approaches that average data across cell populations, single-cell sequencing reveals the unique genomic, epigenomic, transcriptomic or proteomic features of individual cells.
Single-cell sequencing is valuable for a wide range of applications. For example, it can uncover cellular heterogeneity within complex tissues – such as tumors – and provide insights into how different cell types or cellular states function, interact and respond to environmental cues or disease processes.
This blog explains what single-cell sequencing is, describes how it works, provides examples of applications, and helps you to decide when to choose single-cell over bulk sequencing.
Table of contents
What is single-cell sequencing?
Single-cell sequencing is a rapidly growing field, and includes a family of techniques designed to isolate and analyze genetic, epigenetic, transcriptomic or proteomic material information from individual cells. The first single-cell sequencing study – which sequenced the mRNA molecules of a manually picked single mouse cell1 – was published by Tang et al. in 2009, and the method has become more and more popular ever since. Today, more than a third of all RNA sequencing publications use single-cell instead of bulk sequencing.2
Traditional bulk sequencing can mask differences between cells by taking population averages, whereas single-cell sequencing provides information about cell-to-cell variability. A good analogy is to compare the different techniques to a fruit smoothie and a fruit salad. In a smoothie, all the ingredients are blended together, giving you a combined flavor profile. Similarly, traditional bulk sequencing analyzes a mixture of cells, providing average molecular data about the original sample. Single-cell sequencing, on the other hand, allows you to analyze each cell individually, and can be compared to a fruit salad, where you can easily tell which flavor is coming from what fruit, and determine the amount of each fruit type included in the salad.
How single-cell sequencing works
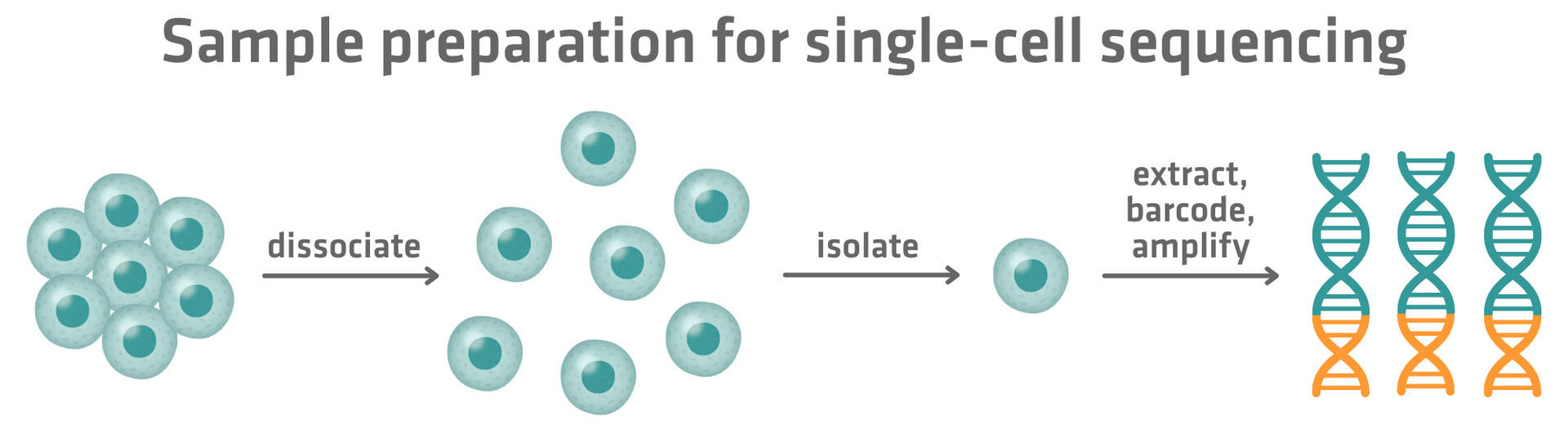
Single-cell sequencing follows a similar workflow to traditional bulk sequencing, consisting of sample preparation, library preparation, sequencing and data analysis. However, it incorporates key steps to dissociate biological samples into single-cell suspensions, to isolate single cells and to handle and track the molecular material from individual cells.3
Sample preparation
The process begins by turning your biological sample into a single-cell suspension. This is achieved by dissociating cells through a combination of mechanical and enzymatic methods. Commonly used mechanical dissociation and enzymatic digestion methods include:
| Mechanical dissociation | Enzymatic digestion |
| Mincing with a scalpel | Typsin/TrypLE™ |
| Dounce homogenization | Collagenase |
| Scraping/aspirating and dispensing with a pipette | Elastase |
| Bead beating/milling | Papain |
| Subtilisin A |
The dissociated cells can then be optionally filtered to eliminate clumps and prevent clogging that may negatively impact single cell isolation later on.
Once a single-cell suspension has been prepared, the sample will contain a mixed population of cells. Before moving on, you might want to sort, enrich or deplete cells to improve the quality and relevance of the sequencing data.
- Sorting
means isolating a specific cell population of interest, ensuring that downstream sequencing focuses only on relevant cell types. - Enriching
means increasing the proportion of desired cells, which is especially useful when the target population is rare. - Depleting
means removing unwanted cells that could dilute or obscure important signals.
Commonly used methods to sort, enrich or deplete cells are fluorescence-activated cell sorting (FACS), magnetic-activated cell sorting, and density gradient centrifugation. For example, LevitasBio offers devices that can enrich samples by depleting dead cells, and the company recommends using our ASSIST PLUS pipetting robot for automated sample preparation.
The cells within the suspension then need to be separated from one another. This isolation can be done by distributing cells into individual wells of a microplate, by sorting them into microwells on a chip, or by encapsulating them into droplets via a microfluidic system. More in-depth information on how plate-, microwell- and droplet-based single-cell sequencing methods work can be found in our article 'Single-cell RNA sequencing methods: plate-, droplet- and microwell-based approaches'.
Once isolated, the molecular material from each cell is extracted and amplified, as the quantity from a single cell is typically too small for sequencing. To avoid the high cost of sequencing each cell individually, a unique molecular barcode is added to the DNA or RNA or proteins from each cell. This barcode acts like an ID tag, allowing all the material from different cells to be pooled into a single sequencing library while still being traceable back to its cell of origin.
Library preparation
The pooled library with the barcoded material from the different cells subsequently needs to be prepared for processing by a sequencing instrument. This usually involves a second round of barcoding, adding sequencing platform-specific adapters to all the nucleic acid or protein fragments.
Sequencing
Once prepared, the pooled library is sequenced. The same sequencing platforms used for bulk sequencing are also used for this step, and Illumina sequencing is the most commonly employed technique. If you would like to learn more on Illumina sequencing and the principles of other sequencing methods, see the following blog posts:
- DNA sequencing methods: from Sanger to NGS
- Sanger sequencing vs NGS
- Short read vs long read sequencing
Data analysis
After sequencing, bioinformatics tools use the barcodes added during sample preparation to deconvolute the data and reconstruct the molecular profile of each individual cell. This enables downstream analysis such as discovering mutations, identifying cell types, or comparing gene or protein expression patterns across a tissue.
Single-cell sequencing datasets are often represented in plots as shown below (Figure 2), with the most important differentiating factors used for the x and y axes. Several dimensionality reduction techniques – like t-SNE (t-distributed stochastic neighbor embedding) and PCA (principal component analysis) – are available to create these plots, which minimize complexity and allow better visualization of the data. Each of the dots in the plots represents a cell, and shows how it is characterized by the main factors. The more similar cells are, the closer together they will be on the plot. You can gain a deep understanding of what connects or differentiates cell types and states by examining what distinguishes the clusters and cells within a cluster from each other.
Each cell cluster is displayed using a different color in Figure 2a. However, sometimes you already know what cell type each cell belongs to, for example because you had FACS sorted them. In this case, you can use single-cell sequencing for outlier clustering. Simply assign a color to each cell type – green for T cells, orange for B cells, etc. – and check if some cells from a certain cluster type are with cells from another type as seen in Figure 2b.
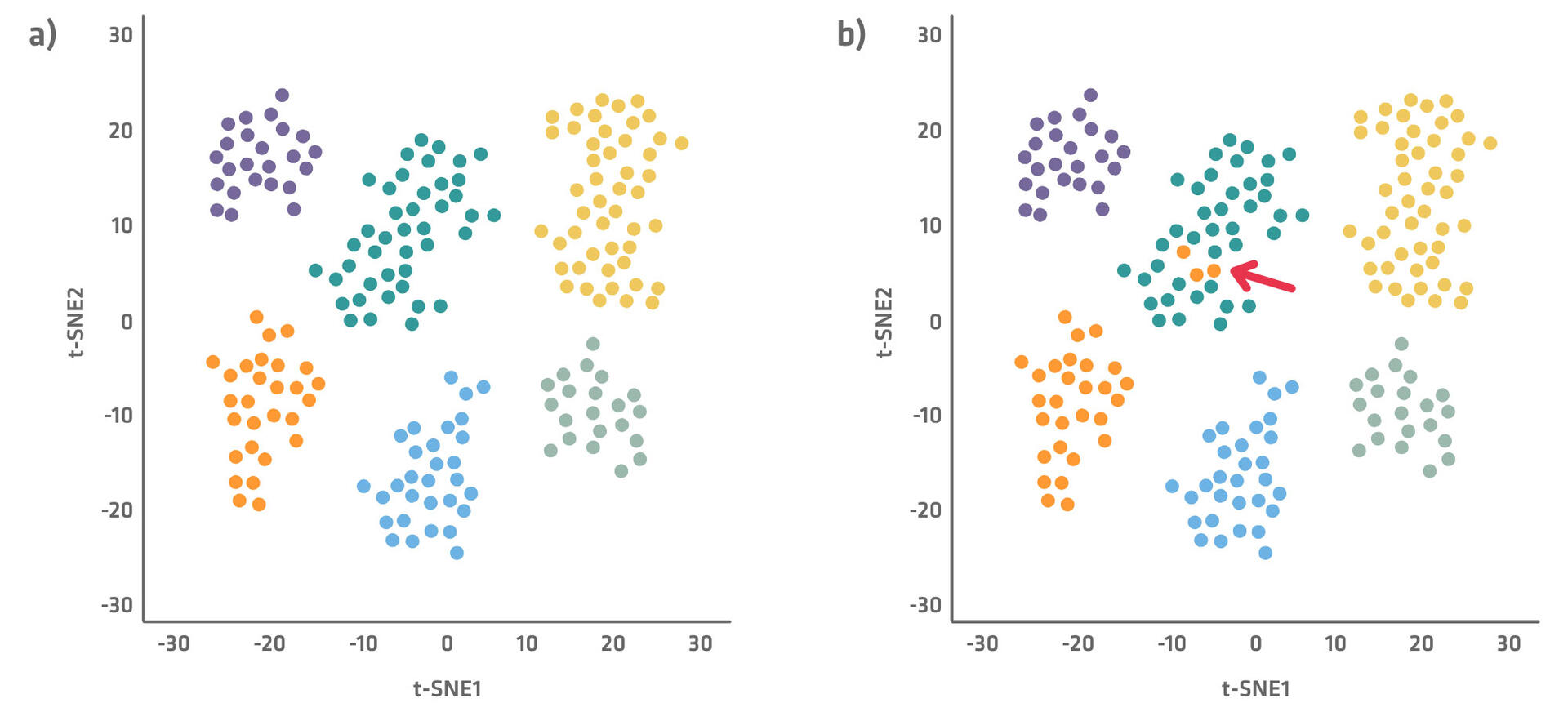
Note that flow cytometry and mass cytometry can also be used to measure heterogeneity across individual cells. However, they are limited to assessing a few dozen pre-selected markers, whereas single-cell sequencing can profile all genes present in a cell, providing a much richer view.
Single-cell sequencing applications
The development of single-cell sequencing has enabled numerous applications across various fields.
Sequencing the genome of different cells can be useful in oncology. Tumors are genetically diverse, and even within the same tumor mass, different cells may carry distinct mutations or copy number variations that can influence how a tumor grows, spreads and responds to treatment. By revealing the cellular heterogeneity and composition of the tumor using single-cell sequencing, personalized and targeted therapeutic approaches can be provided. Single-cell genome sequencing is also commonly applied in microbiome and environmental analyses, as it enables researchers to determine the composition of complex microbial communities by isolating and sequencing the genome of individual cells without culturing them. This is particularly valuable for microbial samples containing unculturable organisms.
Sequencing the epigenome or transcriptome or proteome of individual cells is useful for studying cellular diversity and regulation in complex systems and tissues. For example, single-cell epigenetic or transcriptomic or proteomic data can reveal the identity, state and function of various immune cell types, helping researchers understand immune responses or dysfunctions. In cancer, these techniques can uncover the epigenetic modifications and gene or protein expression patterns that distinguish different cell populations in the tumor microenvironment. They are also widely used in developmental biology to trace how stem cells differentiate into specialized cells, and in disease research to identify cell-specific markers, drug targets or regulatory changes that underlie disease.
Multi-omics applications that sequence several of these molecular layers at once are also available. For example, single-cell nucleosome, methylome and transcriptome sequencing (scNMT-seq) offers simultaneous insights into nucleosome occupancy, methylome and transcriptome at a single-cell level. If you would like to learn more about how this works, refer to our article 'Efficient high throughput scNMT profiling with the VIAFLO 384'.
Another example of a multi-omics single-cell sequencing approach is the ResolveOME technology from BioSkryb Genomics, which can be used to amplify both the genome and transcriptome within each cell.
Pros and cons of single-cell sequencing
Single-cell sequencing has many advantages over more traditional bulk sequencing, but there are limitations too.
Bulk sequencing is generally cheaper and faster than single-cell approaches. The protocols are simpler, allowing you to achieve a higher throughput with a lower budget and within a shorter time frame. In addition, pre-processing cell culture or tissue biopsy samples is more straightforward for bulk methods – as they don't require single-cell isolation – further simplifying the workflow. Moreover, bulk sequencing has been used for decades, and is still the more commonly used method, meaning its protocols are established and robust, making them reliable for generating reproducible results.
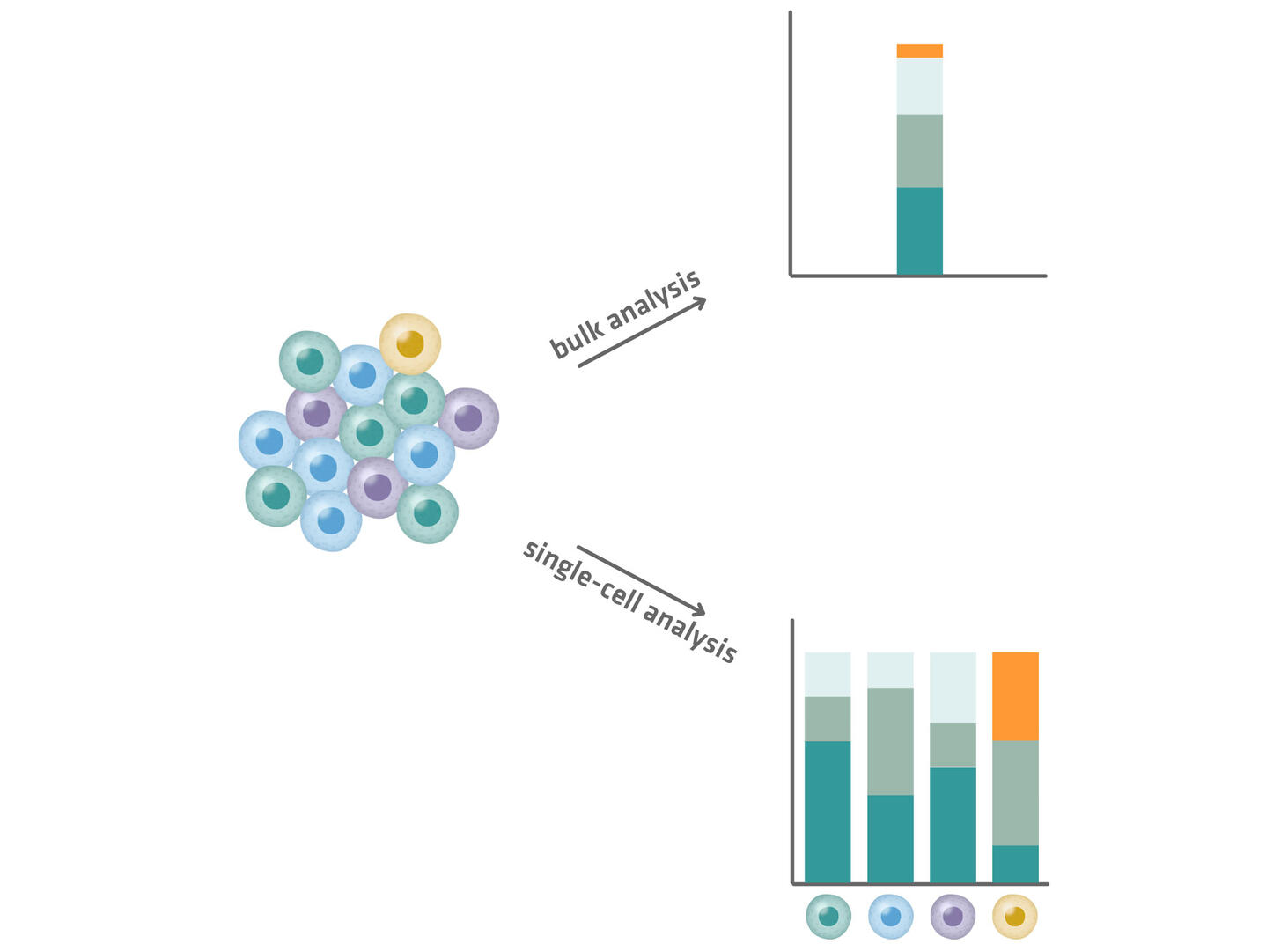
However, information from individual cells is lost in bulk sequencing, and information from rare molecular species is diluted in the average signal of the entire cell population, as shown in Figure 3. In a bulk RNA sequencing experiment, a small fraction of transcripts (orange segment in Figure 3 below) may appear insignificant due to a low abundance. In a single-cell experiment, these transcripts might be revealed as critical markers of a rare but significant cell population (yellow cells in Figure 3 below). The yellow cells could, for example, be cancer stem cells from a tumor sample, and the orange segments could encode a resistance factor to certain therapies, which is something that only single-cell sequencing can reveal.
Single-cell approaches are also better suited to spatial transcriptomic analyses where the goal is to correlate transcriptional profiles with the physical location within the tissue’s original architecture.
In bulk sequencing, spatial information is largely lost. Researchers can slice a tissue, isolate a small region of interest, and sequence it as a homogenized sample to obtain gene expression data that can be attributed to a certain tissue region, however they cannot create a fine-grained map of where specific transcripts originated. In contrast, single-cell spatial transcriptomics methods preserve positional context. One option is to place a tissue section onto a slide patterned with barcoded capture spots. Each spot captures mRNA molecules from the overlying cells, tags them with a unique positional barcode, and reverse transcribes them. After sequencing, these barcodes allow each read to be traced back to its precise location on the slide, creating a high resolution spatial map of gene expression across the tissue.
Conclusion
Single-cell sequencing represents a transformative leap from traditional bulk sequencing approaches, offering unprecedented insights into cellular diversity and function. Bulk sequencing analyzes pooled data from many cells and provides average signals that can mask rare and unique cell types, while single-cell sequencing analyzes individual cells separately to reveal cell-to-cell variability and identify even the rarest cellular populations. This fundamental difference makes single-cell approaches particularly valuable for detailed cellular insights, cell lineage tracking and differentiation studies that would be impossible with bulk methods.
The choice between bulk and single-cell sequencing ultimately depends on your research objectives and resources. Bulk sequencing remains an excellent choice for general profiling applications where population-level averages are sufficient, offering a simpler, more affordable workflow with established protocols. However, when your research requires high resolution at the single-cell level, sensitivity to cellular heterogeneity, or spatial transcriptomic analyses with preserved positional information, single-cell sequencing becomes indispensable despite its increased complexity and cost.
| Bulk sequencing | Single-cell sequencing |
| Analyzes pooled data from many cells | Analyzes individual cells separately |
| Provides average signal | Reveals cell-to-cell variability |
| Masks rare and unique cell types | Identifies rare and unique cell types |
| Used for general profiling | Used for detailed cellular insights |
| Cannot track cell lineage | Can track cell lineage and differentiation |
| Workflow is simpler and more affordable | Workflow is more costly and complex |
| Limited resolution | High resolution at single-cell level |
| Less sensitive to heterogeneity | Highly sensitive to heterogeneity |
| Spatial information is largely lost | Ideal for spatial analyses |
Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.