
An introduction to the different types of ELISA tests
Written by Éva Mészáros
14. September 2022
ELISA stands for 'enzyme-linked immunosorbent assay’, and ELISA tests are mainly used in research and clinical applications, including detecting viral infections such as hepatitis B virus (HBV), hepatitis C virus (HCV) or human immunodeficiency virus (HIV). In this article, we will outline the different types of ELISAs, explain how to analyze the assay data, and give an overview of the reagents and equipment required.
Table of contents
What is an ELISA?
Let's start with a general definition of an ELISA before looking at different assay methods:
ELISA
An ELISA test is a type of enzyme immunoassay (EIA). These highly specific and sensitive assays are used to detect concentrations as low as 0.01 nanograms of antigen or antibody per milliliter. When performing ELISAs, antigen-antibody complexes are immobilized to a solid surface. An enzyme is covalently attached to one of the molecules in the complex, and the subsequent addition of an enzyme-specific substrate results in a colored reaction product.
Different types of ELISA
The four main types of ELISAs are direct, indirect, sandwich and competitive. All of these tests are commonly performed in 96 well plates, using the bottom of the wells as the solid surface to immobilize the antigen-antibody complexes. The differences between the four types – and their advantages and disadvantages – are discussed in the following sections.
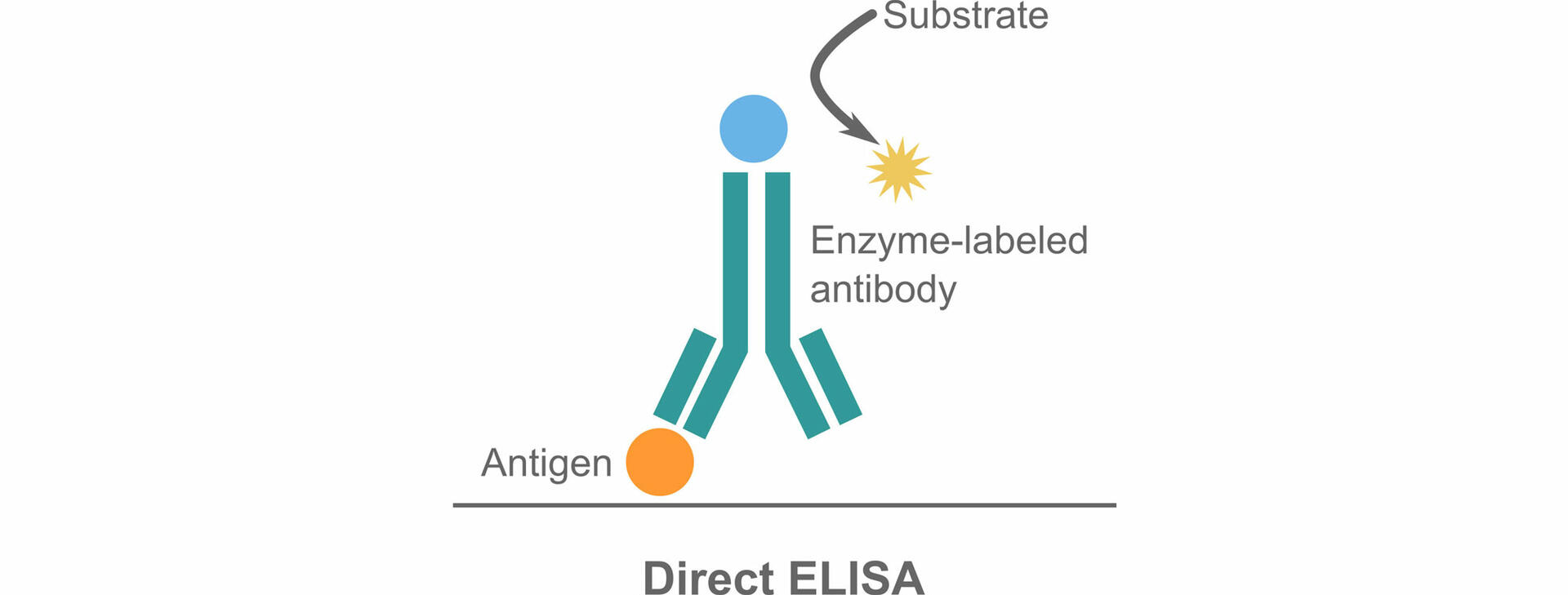
Direct ELISAs
The direct ELISA protocol can be used to detect antigens, and consists of four steps:
- Plate coating: Dilute your samples using a coating buffer, then pipette them into a microwell plate. After incubation, discard the coating solution and wash the plate with a wash buffer. The proteins contained in your sample – including the antigen of interest – are now immobilized to the plate.
- Plate blocking: Dispense blocking buffer into the plate and incubate it. The blocking buffer will bind to any remaining protein-binding sites in the coated wells, reducing subsequent non-specific binding of antibodies to the plate. Wash your plate again.
- Antibody incubation: Transfer enzyme-labeled antibodies to the plate, then incubate. The antibodies will bind to the antigens of interest in wells that contain the analyte. Following incubation, wash away unbound antibodies with a wash buffer.
- Detection: Add an enzyme-specific substrate to the plate. The enzymes covalently attached to the antibodies will start producing a colored reaction product. After adding a stop solution to terminate the color development, read the absorbance of each well with a plate reader. The signal intensity allows you to determine whether a sample contains the antigen of interest, and at what concentration. More information on how to analyze ELISA data can be found below.
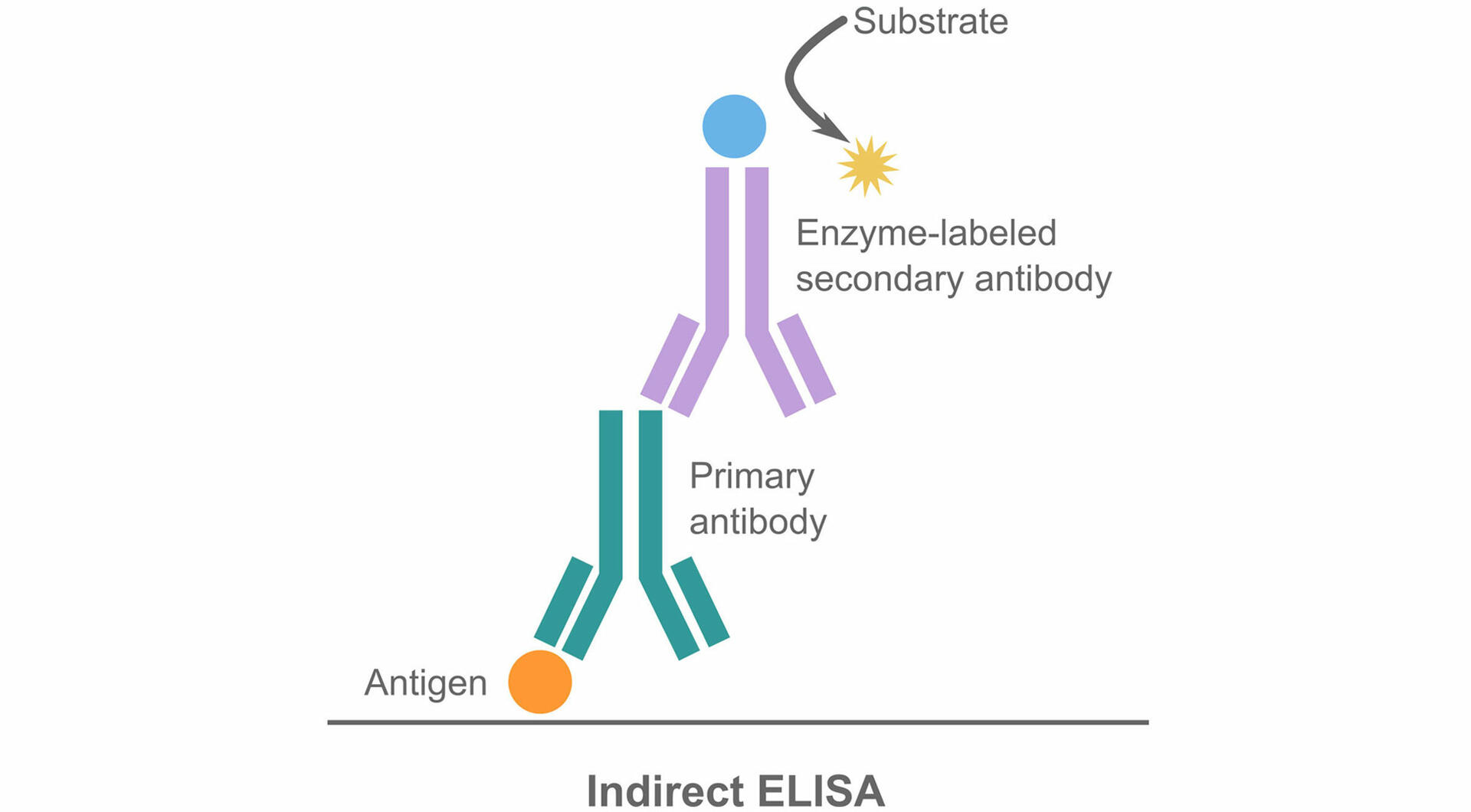
Indirect ELISAs
The only difference between a direct and indirect ELISA is that the antibody incubation process is divided into two steps. After coating and blocking the plate, primary antibodies that bind to the antigens of interest are added to the plate, which is then incubated. Following a wash step, enzyme-conjugated secondary antibodies – which bind to the primary antibodies but not to the target antigens – are added. After incubating the plate a second time and washing off unbound secondary antibodies, the detection step can be performed.
Direct vs. indirect ELISAs
The advantage of direct ELISAs is that they save time and reagents, as the protocol is slightly shorter. In addition, the risk of cross-reactivity is reduced. Cross-reactivity occurs when antibodies bind non-specifically to epitopes on molecules that are not the antigen of interest. Because direct ELISAs use only one antibody instead of two, this risk is lower.
Despite the longer protocol and higher risk of non-specific binding due to cross-reactivity, indirect ELISAs have many advantages too. For example, they are more sensitive, because multiple secondary antibodies can bind to one primary antibody, amplifying the detectable signal.
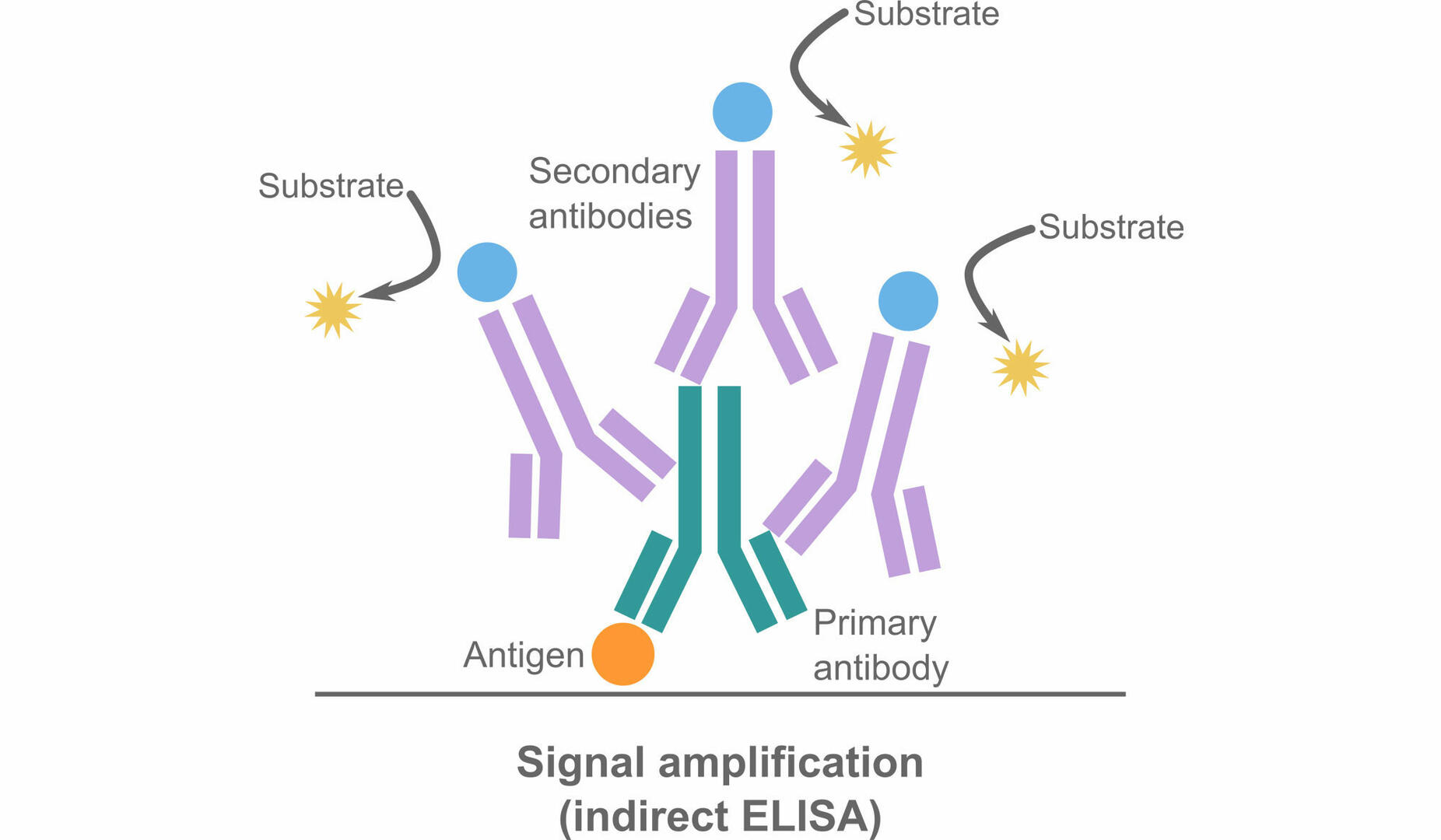
Moreover, indirect ELISAs offer greater flexibility. A secondary antibody can be specific to several primary antibodies, as long as they are the same type and from the same host species. This means that the same labeled secondary antibody can be used in different indirect ELISA applications, whereas several labeled primary antibodies are needed to perform different direct ELISA protocols.
Another advantage of indirect ELISAs is that they can not only detect antigens, but also antibodies. This requires coating the plate with antigens that bind to the antibodies of interest, then adding the samples in place of the primary antibodies during the first antibody incubation step.
Sandwich ELISAs
For sandwich ELISAs, the plate isn't coated with antigens, but with capture antibodies specific to the antigen of interest. After blocking the remaining protein binding sites with a buffer that is designed to bind to the sites not occupied by the capture antibodies, the samples can be added to the wells. During incubation, the antigens of interest bind to the capture antibodies. Next, the plate is incubated either with enzyme-labeled detection antibodies (direct sandwich ELISA) or with primary antibodies followed by enzyme-labeled secondary antibodies (indirect sandwich ELISA). Please note that the capture and primary antibodies for indirect sandwich ELISAs need to be from different host species to increase specificity.
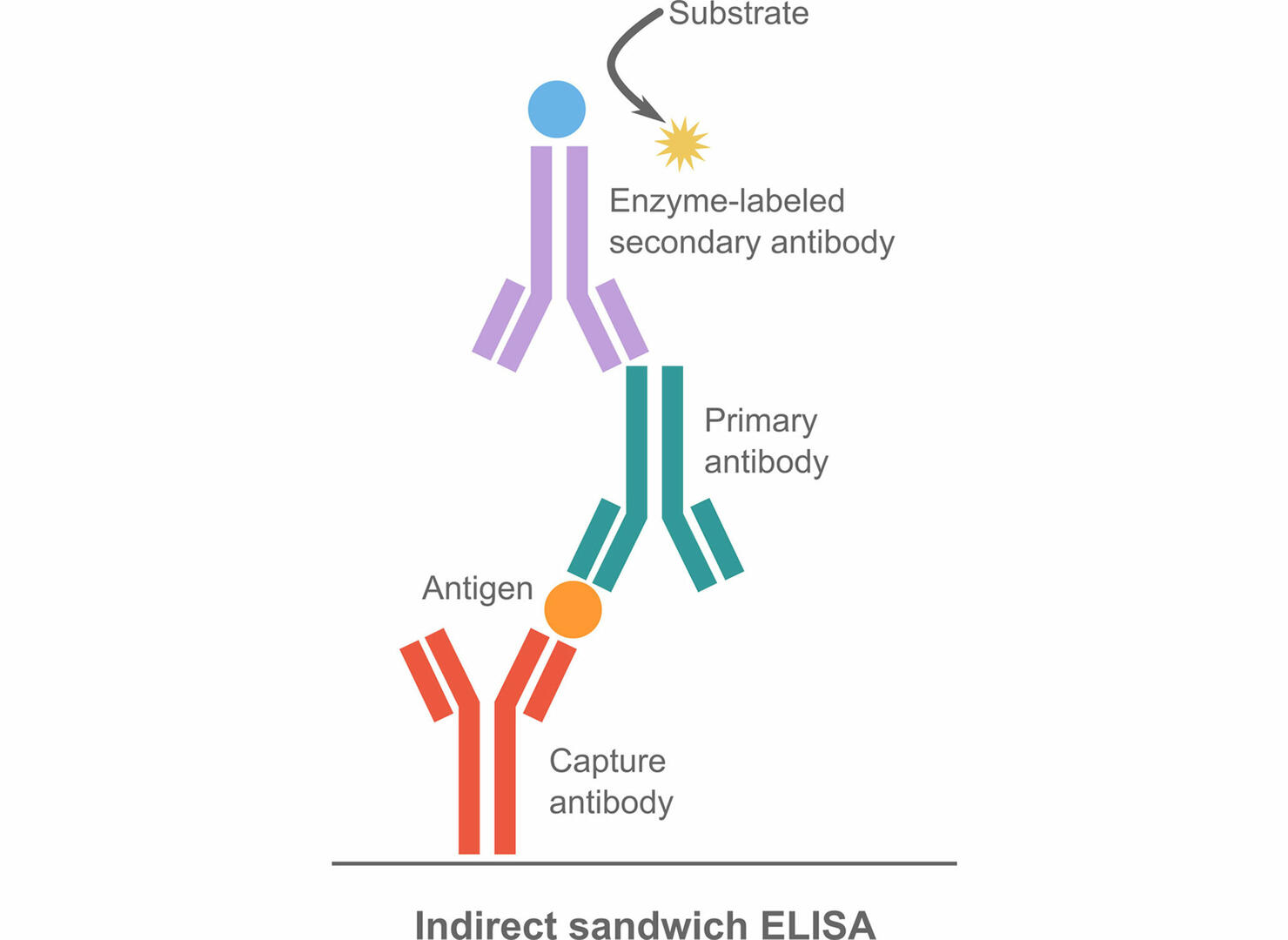
The advantage of sandwich ELISAs is that they are highly specific. The two antibodies capturing an antigen bind to different epitopes of it, making it almost impossible for both antibodies to bind non-specifically. Thus, sandwich ELISAs are the perfect method for complex samples. A disadvantage of sandwich ELISAs is that it can sometimes be difficult to find two antibodies that work well together while binding to different epitopes of the same antigen.
Competitive ELISAs
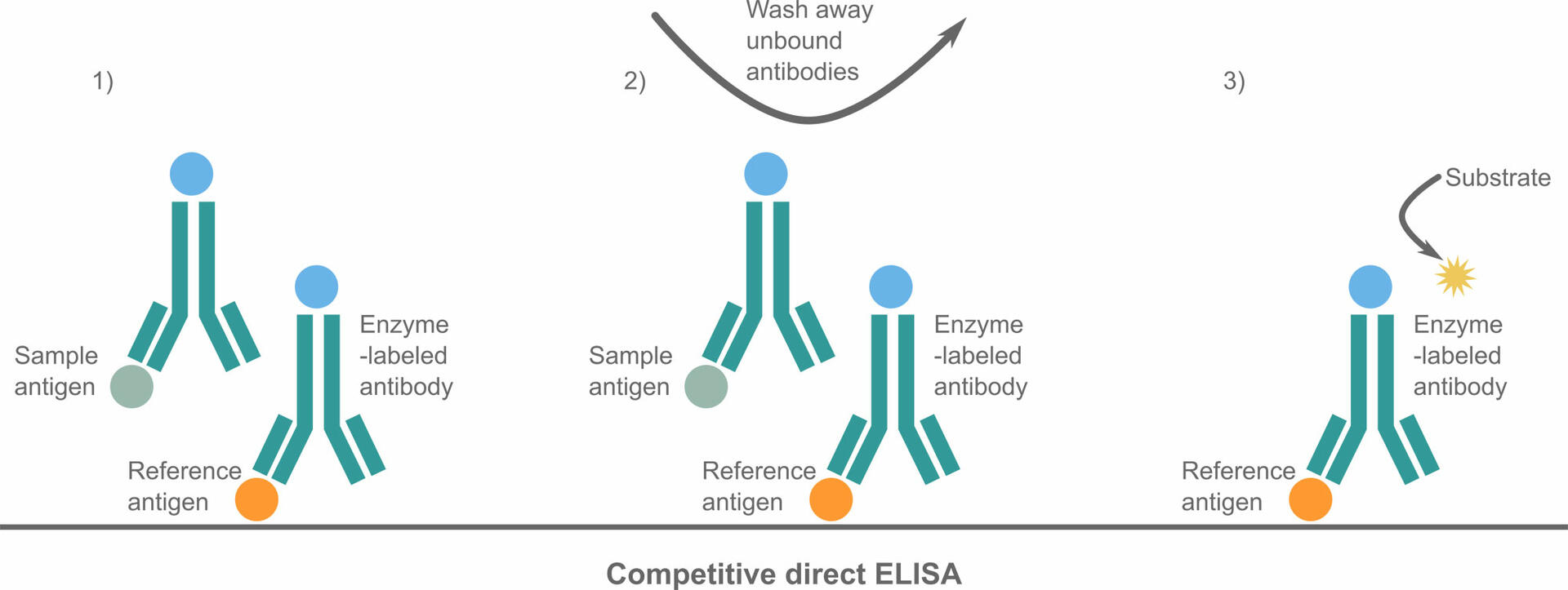
In competitive ELISAs, also known as inhibition ELISAs, the amount of labeled molecules is limited, and the sample antigens or antibodies compete with reference antigens or antibodies to bind to these labeled molecules. This means that the analyte concentration is measured by the detection of signal interference. Direct, indirect and sandwich ELISAs can all be adapted to this format, but the steps and reagents of a competitive ELISA protocol can vary. To perform a competitive direct ELISA, you could, for example, proceed as follows:
- Coat the plate with a reference antigen and block the remaining binding sites with a buffer.
- Incubate your sample that has an unknown antigen concentration with a limited amount of labeled antibodies. If the antigen concentration in the sample is low, a large portion of the antibodies will not be able to bind to an antigen, and vice versa.
- Add the sample-antibody mixture to the coated wells and incubate the plate. The antibodies that couldn't bind to a sample antigen in the previous step will now bind to a reference antigen (step 1 in the image below).
- Wash your plate. Antibodies bound to sample antigens will be washed away because they aren't immobilized to the plate (step 2 in the image below).
- Add a substrate (step 3 in the image below) and measure the colored reaction product. The stronger the color change, the less antigen was present in the sample, and vice versa.
The advantages and disadvantages of a competitive ELISA depend on the base ELISA (direct, indirect, competitive) selected.
How to analyze ELISA results
After looking at the working principle of the different types of ELISAs, let's now talk about data analysis. ELISAs can either deliver qualitative, semi-quantitative or quantitative results:
- Qualitative results: Is the antigen or antibody present in a sample or not?
- Semi-quantitative results: Is the antigen or antibody concentration in sample A higher or lower than in sample B?
- Quantitative results: What is the antigen or antibody concentration in a sample?
To obtain reliable results, you should include negative and positive controls in every ELISA plate that you set up. Negative controls will allow you to check for false positive results caused by non-specific binding or contamination, whereas positive controls confirm that the test is working as intended, even if all the samples are negative.
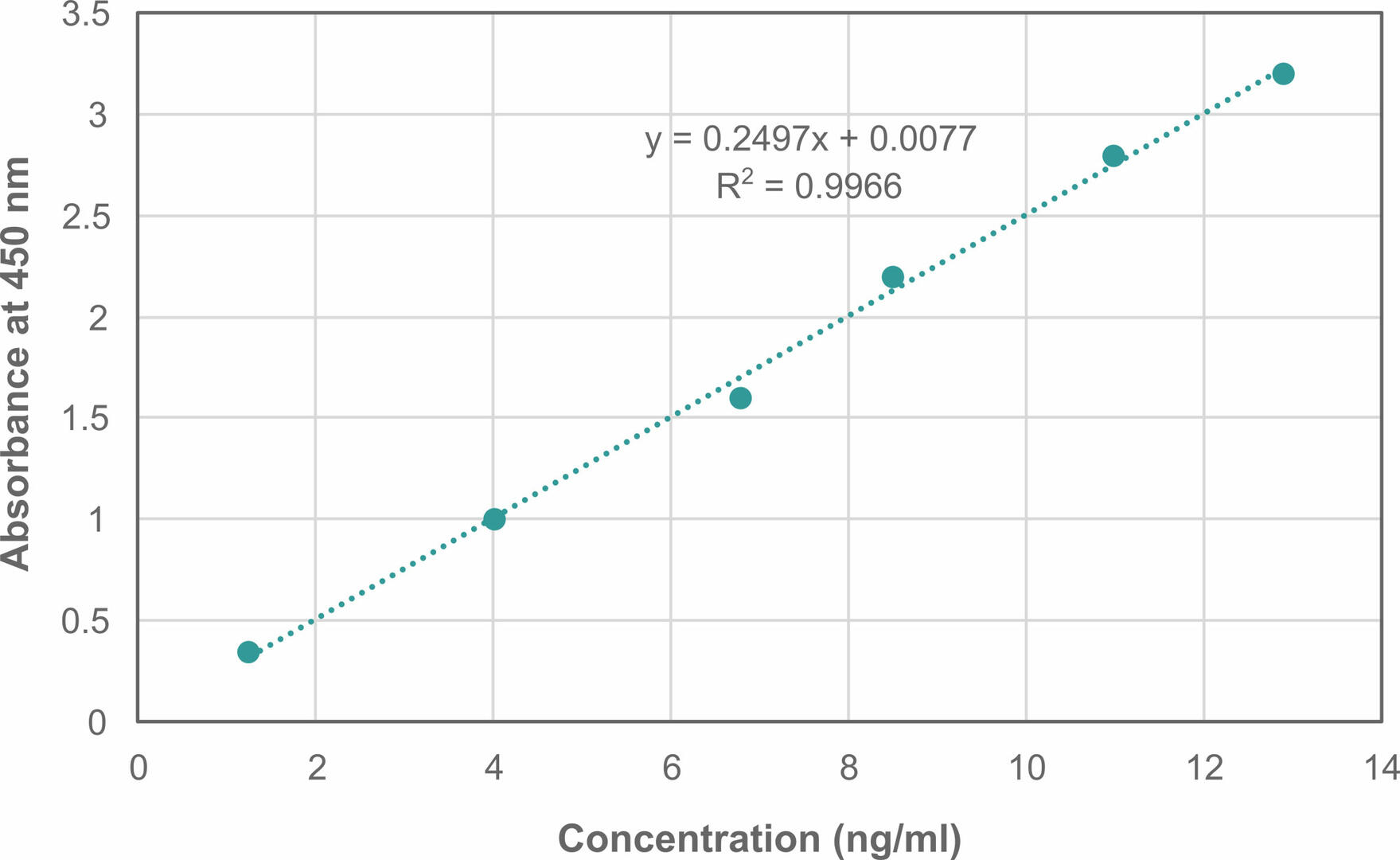
If you want to obtain quantitative results, you also need to set up a standard curve. To do so, serially dilute a sample with a known amount of the antigen or antibody of interest with a diluent buffer. After running the ELISA, plot the known concentrations against the obtained absorbance values, using curve fitting and data analysis software to find the curve that best fits your data and give you an equation to calculate the unknown antigen or antibody concentrations in your samples. For example, if a linear plot is the curve that fits your data best, the standard curve will show the concentration on the x-axis and the absorbance values on the y-axis,1,2 with R2 indicating how closely the data points fit the trendline. Values greater than 0.99 allow you to get accurate results.1,3
The equation for the linear regression line of the standard curve (y = mx +b) will then allow you to calculate the antigen or antibody concentration of your samples. As y corresponds to the absorbance and x to the concentration, the equation for the linear regression line is equivalent to:
Absorbance = m(concentration) + b
Solving this equation for the concentration will give you the formula:
Concentration = (Absorbance - b) / m
For example, if your equation for the linear regression line is y = 0.2497x + 0.0077 (as in the image above), a sample with an absorbance value of 2 would have a concentration of 7.9788:
7.9788 = (2 - 0.0077) / 0.2497
When performing quantitative ELISAs with complex samples, you should also include a spike control. A spike control is a serial dilution of a sample with a known amount of antigen or antibody of interest in serum instead of diluent buffer. Comparing the absorbance values of the spike control and the standard curve will allow you to see whether proteins other than the antigens or antibodies of interest in your sample hinder antigen-antibody binding, leading to an underestimation of the target concentration.
Note that the negative and positive controls, standard curves, spike controls and samples should all be run in duplicate or triplicate. This lowers the number of samples that can be analyzed per plate, but increases assay reliability. The chance that factors such as pipetting errors remain unnoticed is much lower when working with average absorbance values of duplicates or triplicates during data analysis, as you would notice high deviations of the mean if only one of the wells in the set was affected. An acceptable deviation of the mean for duplicates is a value of 20 % or lower.2
Reagents for ELISAs
The reagents needed for an ELISA test depend on the protocol, but generally include coating, blocking and wash buffers, enzyme-conjugated antibodies or antigens, a substrate and a stop solution. For the detection of common antigens and antibodies, you can usually purchase ready-to-use ELISA kits that contain everything you need.
The most widely used reagents in these kits are:
- Coating buffer: The two most common coating buffers are PBS and bicarbonate. They stabilize the antigen or antibody used to coat the plate during incubation.4
- Blocking buffer: To prevent non-specific binding of detection antibodies to the plate surface, remaining protein binding sites need to be blocked. This is achieved with blocking proteins such as bovine serum albumin (BSA), non-fat dry milk (NFDM), casein or caseinate, normal serum or fish gelatin.5
- Wash buffer: Wash steps to remove unbound materials are mostly performed with PBS containing a small concentration of a nonionic detergent, such as Tween® 20.4
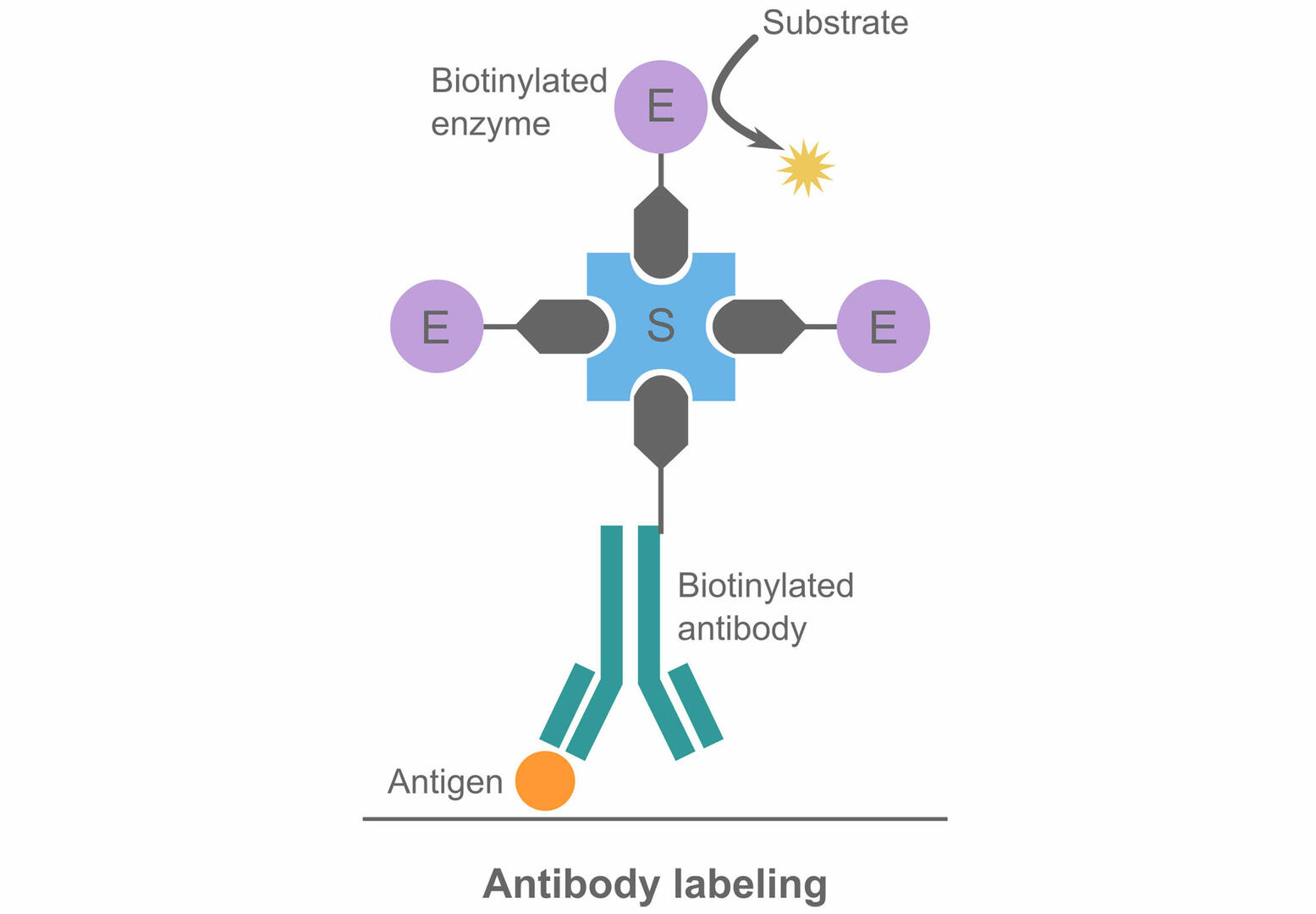
- Enzyme-conjugated antibodies or antigens: The antibodies or antigens required will depend on the analyte. To label the antibodies or antigens with an enzyme, a streptavidin-biotin bridge is generally used to link the antibodies or antigens to the detection enzyme.6 The two most common enzymes used are horse radish peroxidase (HRP) and alkaline phosphatase (ALP).7
- Substrate and stop solution: The substrates and stop solutions are enzyme specific. The substrate for HRP contains TMB (3,3′,5,5′-tetramethylbenzidine), substrate buffer and hydrogen peroxide, and the reaction can be stopped with sulfuric acid. The substrate for ALP is a p-nitrophenyl phosphate (pNPP) solution,4 and the reaction can be stopped with NaOH.8
Equipment for ELISA
To perform an ELISA, you need pipettes to add the reagents and samples to your microwell plate, an incubator to hold it at a constant temperature, a plate washer to remove unbound molecules, and a plate reader for assay analysis.
If you're working in a lab with a limited budget or low throughput, there might be no plate washer at hand. In this case, you can also use pipettes in combination with an aspiration system for the wash steps. During the COVID-19 pandemic, for example, our VACUSIP aspiration systems have been used for sandwich ELISAs detecting antibodies against the SARS-CoV-2 virus in patients that have recovered.
On the other hand, high throughput labs might be looking for solutions to increase their productivity. In this case, various pipetting solutions may come in handy, including adjustable tip spacing pipettes to transfer samples between different labware formats, 96 channel pipettes to simultaneously add reagents to every well, or small benchtop pipetting robots to automate workflows. If you would like to learn more about the solutions we offer, read the articles below:
Adjustable tip spacing pipette
96 channel pipette
- VIAFLO 96 channel pipette increases the throughput of ELISA, Luminex and flow cytometry workflows at the University of North Texas
- Application note: Performing an ELISA with the VIAFLO 96/384 handheld electronic pipette
Pipetting robot
- ASSIST PLUS pipetting robot automates HLA typing workflows
- Application note: Performing an ELISA with the ASSIST PLUS pipetting robot
96 channel pipette and pipetting robot
Conclusion
ELISAs have been used since the 1970s, and are considered the gold standard of immunoassays.9,10,11 We hope that this article has helped you to get an overview of the most relevant aspects related to this important application.
Did you like this article?
Further reading:



