DNA purification: comparing different methods and techniques
Written by Éva Mészáros · 30. May 2024
This article was originally published on 16. July 2021 and has since been updated.

Purifying DNA is a common process in molecular biology. Unlike DNA extraction, it doesn't include any lysis steps to break the cell membrane and liberate the DNA. Instead, it involves the clean-up of your samples, e.g. to effectively remove all components that were used to facilitate amplification of the target sequence during a polymerase chain reaction (PCR).
Various DNA purification methods are available and this article will provide a detailed comparison of their advantages, disadvantages, and applications. Read on to learn how to ensure that your samples are pure enough for your downstream application. Please note that most of the techniques explained in this article can also be used if you want to purify RNA instead of DNA.
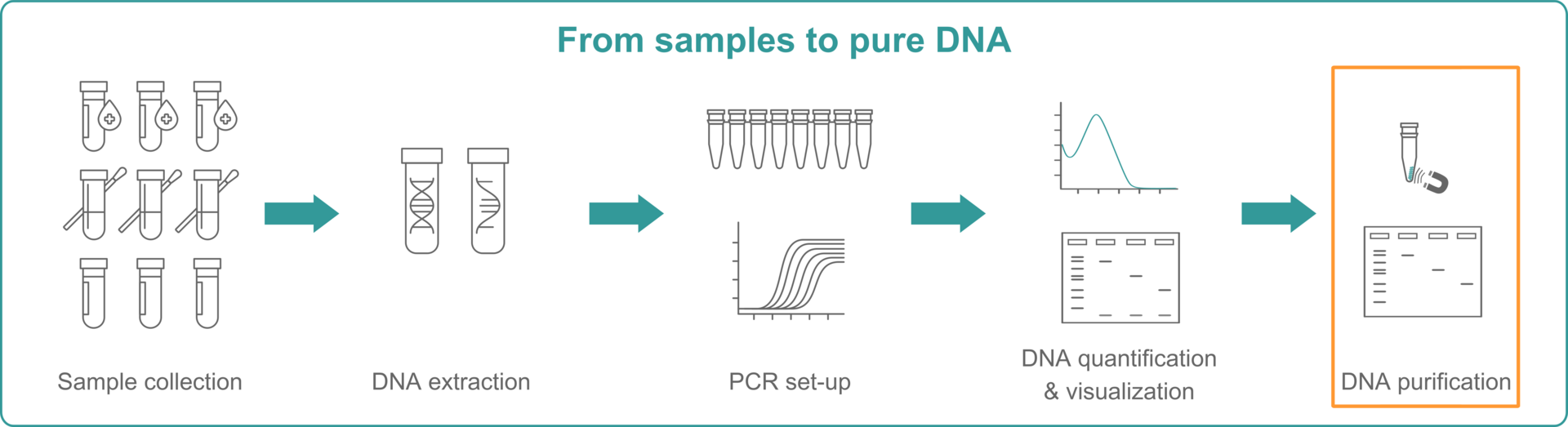
This article represents the fifth and last part of our blog series covering how to get from samples to pure DNA.
Table of contents
Ethanol and isopropanol precipitation
The first purification method we'll have a closer look at is ethanol and isopropanol precipitation. This is the method of choice for purifying genomic DNA, and can be used to concentrate and de-salt samples after applications such as CsCl density gradient centrifugations with EtBr, phenol-chloroform extractions, digestions and PCRs.
How does it work?
Ethanol and isopropanol precipitation are all about solubility. Water and DNA are both polar, which is why DNA molecules dissolve easily in water. To precipitate them, you can either use ethanol or isopropanol.
For ethanol precipitation, you need to add twice the sample volume of ice-cold 96 percent ethanol, and salt (commonly sodium acetate) to the solution. Ethanol lowers the dielectric constant, allowing the negative charges on the sugar-phosphate backbone to be neutralized by the Na+ ions of sodium acetate. Since the DNA molecules are now less hydrophilic, they will drop out of the solution when you incubate the mixture on ice. Then, centrifuge your sample to separate the DNA from the rest, and wash the pellet in cold 70 percent ethanol to remove any residual salt. Centrifuge the sample a second time, remove the ethanol, allow the DNA pellet to dry, and resuspend it in a clean aqueous buffer.1 To dry the pellet, you can either place the tube (with the lid open) in a laminar flow hood for several hours, or use a vacuum centrifuge. The method you choose is up to you, but you have to ensure that the pellet is completely dry to avoid residual ethanol negatively affecting your downstream applications.
Isopropanol precipitation is very similar. The only differences are that you can skip the incubation on ice step, and replace ice-cold ethanol with room-temperature isopropanol for the first step. Regarding the volume of isopropanol, an amount equal to the sample volume is sufficient.2
Whether ethanol or isopropanol is more suitable depends on your sample volume, concentration and the size of your DNA fragments. If you have a large sample volume, it may be impossible to add twice the sample volume of ethanol into the tube. Isopropanol precipitation is also preferable for the precipitation of larger DNA fragments and lower sample concentrations, as DNA is less soluble in isopropanol. On top of that, isopropanol precipitation is the faster method as you don't need to incubate your samples before centrifugation.2
Advantages and disadvantages
Ethanol and isopropanol precipitation aren't costly at all, as ethanol, isopropanol and sodium acetate are very affordable, and the process provides a good yield of high purity DNA. They are, however, very time-consuming processes and, as they need to be performed manually, are highly variable. Consequently, low reproducibility can be a problem.
Equipment needed
All you need for ethanol or isopropanol precipitation is a centrifuge and a laminar flow hood, or a vacuum centrifuge to dry the pellet.
Gel electrophoresis
As gel electrophoresis separates DNA fragments based on their length, you can use this method to separate sequences of interest from other nucleic acid types and contaminants.
How does it work?
First of all, you need to run a gel. More detailed information on how to perform agarose gel electrophoresis can be found in our article on DNA quantification.

After visualizing your gel, use a sharp scalpel to excise the DNA band of interest. Always remember to wear appropriate personal protective equipment (PPE), such as a face shield and gloves, for this step, especially when using UV light for the visualization of your bands. Then, purify your DNA bands from the TAE- or TBE-buffered agarose gel by using a suitable spin column purification kit. As the various kits available differ slightly from one another, you should carefully follow the manufacturer's instructions. Usually, you need to weigh the DNA bands, add a specified amount of buffer for every 100 mg of gel slice, and heat the mixture to solubilize the agarose.3,4 You then transfer your samples to the spin columns and purify the DNA using binding, washing and elution steps. More detailed information on how spin column protocols work can be found in our article on DNA extraction.
Advantages and disadvantages
The huge advantage of this clean-up method is that agarose doesn't denature the DNA fragments, making it easy to recover them from the gel without any damage. It is, however, not suitable for high throughput labs, because running a gel is very time consuming, and is limited to a low number of samples.
Equipment needed
Compared to other purification methods, you need a lot of different instruments for this workflow. Agarose gel electrophoresis requires a gel electrophoresis system, an external power supply and a biosafety cabinet, as you'll be working with hazardous intercalating dyes. Spin column purification is less demanding, as you only need a centrifuge.
Spin column purification
Spin columns are not only used to extract DNA molecules from lysed samples, but also to purify them, e.g. after a PCR reaction to remove salts, enzymes, primers, primer dimers and nucleotides that may inhibit subsequent applications.
How does it work?
As explained above, spin column purification protocols consist of binding, washing and elution steps. After transferring your samples into the spin columns, you centrifuge them to bind the DNA to the membrane inside the column. This allows unwanted components to pass through. Several additional centrifugation steps with a wash buffer remove residual unwanted materials, and a final centrifugation step with an elution buffer liberates the DNA from the membrane.
Advantages and disadvantages
As you can see, this purification method is quick and easy. On top of this, it can be adapted to your sample number, as you could also use 96 well silica membrane plates instead of single spin columns. However, the membrane may get clogged and, as a minimum elution volume of 30-50 μl is required, you may get rather low DNA concentrations.
Equipment needed
The only piece of equipment needed for spin column purification is a centrifuge, unless you work in the 96 well format and prefer to use a vacuum manifold with a pump, which is also possible.
Magnetic bead purification
Just like spin columns, magnetic beads can be used either for the extraction of DNA molecules from lysed samples or for their purification. For example, you can use magnetic beads to remove salts, enzymes, primers, primer dimers and nucleotides from PCR products that would otherwise inhibit subsequent applications.
How does it work?
The purification workflow with magnetic beads is very similar to the extraction workflow. You first add magnetic beads that bind the DNA molecules to your samples.

You then place the tubes on a magnet to remove the supernatant containing unwanted, unbound material. You repeat this step several times, replacing the wash buffer in between. In the end, you add an elution buffer and transfer the samples to a different vessel.
In contrast to extraction protocols with magnetic beads, only DNA fragments of a particular length bind to the beads during purification. This size exclusion mechanism is achieved by creating the perfect binding conditions for the DNA fragments of interest through varying the buffers, salts, and their concentrations, and therefore the hydrophilicity/hydrophobicity.
Advantages and disadvantages
A huge advantage of magnetic bead purification is that the equipment needed can be chosen based on your budget.
- Option 1: Get a magnetic stand and perform the workflow manually. This is the cheapest option, but also the most tedious and error-prone method. You need to be very careful not to aspirate the beads, as this would result in sample loss.
- Option 2: For high throughput applications, you can also buy a benchtop pipetting robot, or a 96 or 384 channel pipette. These devices reduce manual liquid handling steps, increasing productivity and reproducibility, and can also be used for other applications. For more information on how such devices can be used for magnetic bead purification, see our application notes:
- Reliable time-to-result with a high throughput PCR purification protocol using magnetic beads
- Automated DNA size selection for flexible NGS workflow integration
- PCR purification with Beckman Coulter AMPure XP magnetic beads and ASSIST PLUS
- PCR purification with Beckman Coulter AMPure XP magnetic beads and VIAFLO 96
- Automated DNA clean-up using the Zymo Research DNA Clean & Concentrator™ MagBead Kit
- Option 3: Automate the entire workflow by buying a dedicated purification system.
Another convenience of magnetic bead purification is that you can work with lower elution volumes than for spin column purification. It is also easily scalable, as you can use it with 384 well plates.
Equipment needed
As described, you can match the equipment for magnetic bead purification that you purchase to your budget. Either buy a magnetic stand if you have a limited budget, get a benchtop pipetting robot or a 96 or 384 channel pipette if you have more money available, or go with a dedicated purification system.
Sephadex® purification
Sephadex purification can be used to purify DNA from smaller molecules, e.g. primers, nucleotides or dyes.
How does it work?
Sephadex is a gel filtration resin made of dextran crosslinked by epichlorohydrin.5 To prepare the resin, you need to add Sephadex powder to spin columns, rehydrate it with water, and centrifuge the columns to eliminate excess water. Before spinning the columns a second time, add your samples on top of the resin. During centrifugation, larger molecules will easily pass the resin and elute, whereas smaller molecules will get trapped in the pores of the dextran beads. The size exclusion capability of the beads depends on the Sephadex type you choose. For example, Sephadex G-25 Medium5 can be used to purify DNA with a molecular weight of >5000, and G-50 Medium6 is suitable for molecules with a molecular weight of >30000.
To speed up this workflow, opt for spin columns prepacked with Sephadex instead of creating them yourself, or work with a 96 well filter plate.
Advantages and disadvantages
Just like spin column purification, this method can be adapted to your throughput needs, by either performing it in spin columns or in 96 well filter plates. The absence of a molecule-matrix binding step also prevents unnecessary damage to the DNA,7 making it a rather gentle purification method. However, it's quite time consuming, especially when preparing spin columns or filter plates yourself, as Sephadex has to rehydrate for about 3 hours, and can't be automated.
Equipment needed
The only piece of equipment needed for Sephadex purification is a centrifuge.
Enzymatic approaches
Some manufacturers offer enzymatic approaches to clean up your PCR products if your subsequent application – including Sanger sequencing, next generation sequencing or SNP analysis – requires your samples to be free from primers and nucleotides.
How does it work?
Enzymatic approaches consist of only 2 steps. First, you add an enzyme mix to your samples and incubate them for 15 minutes at 37 °C. During incubation, the first enzyme in the mix, exonuclease I, will digest excess primers, and the second enzyme, alkaline phosphatase, will dephosphorylate nucleotides that were not consumed during PCR. Once the enzymes have served their purpose, you can heat up your samples to 80 °C for another 15 minutes to deactivate the enzymes.8,9,10
Advantages and disadvantages
In addition to being quick and easy, enzymatic approaches result in no sample loss, and can easily be adapted to your sample number. Their only disadvantage is that they can only be used to clean up PCR products from primers and nucleotides, and do not remove any other contaminants.
Equipment needed
All you need for enzymatic PCR clean-ups is an incubator.
Conclusion
As you can see, there is a DNA purification method available for every sample type, downstream application and budget. We hope that this article helped you determine which one to choose for your specific needs.
And, if you haven't only come here for tips on DNA purification, you might also be interested in learning more about sample collection, DNA extraction, PCR lab set-up and DNA quantification. We put together this 5-part blog series when we were in the process of equipping a new in-house lab that now looks like this:

Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.