Understanding shotgun sequencing: a comprehensive overview
Written by Éva Mészáros · 13. September 2024
Shotgun sequencing is an approach used to determine the nucleotide sequence of an organism's genome. The process involves breaking the genome into shorter fragments, sequencing these fragments, and then reassembling them in the correct order.1
The fragmentation process of shotgun sequencing is random, like a shotgun blast, from which the name of the method is derived. Any available sequencing method can then be used for the step of sequencing the fragments. Historically, Sanger sequencing was the standard approach, but today, short-read sequencing methods like Illumina sequencing, or long-read sequencing techniques like PacBio and Nanopore sequencing, are usually employed. For a comparison of these methods, refer to this article: Sanger Sequencing vs NGS.
Table of contents
History and development of shotgun sequencing
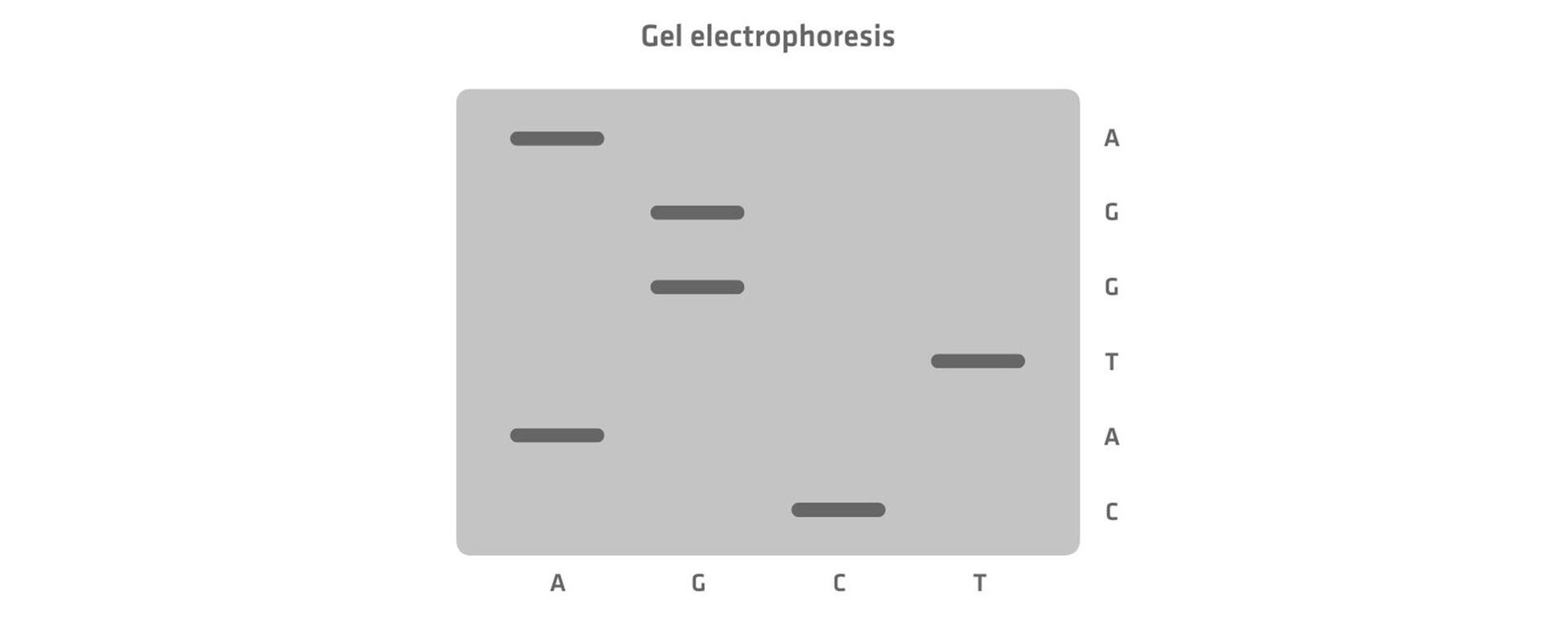
Sanger sequencing, invented in 1977, was one of the first DNA sequencing methods. It involved performing 4 separate PCR reactions per sample, each with normal nucleotides and 1 type of radiolabeled chain-terminating nucleotides. When incorporated into the growing DNA strand, these labeled nucleotides stopped the elongation process, creating DNA fragments of varying lengths. After amplification, the fragments were separated by size using gel electrophoresis, and the sequence was determined by reading the position of the radiolabeled bands. In the image below, the DNA segment travelling furthest produced a band in lane C, which means that the first nucleotide would be cytosine, and so on.2 For more details on Sanger sequencing, see DNA sequencing methods: from Sanger to NGS.
A major limitation of Sanger sequencing was that its read length was limited; very few long fragments were produced, since chain termination eventually occurred. While one could have sequenced as far as possible to then design a new primer for the subsequent segment, a more efficient method was needed.3
In 1979, Roger Staden proposed shotgun sequencing as a solution. This approach involved fragmenting an organism's genome into random sequences, sequencing them in parallel, and reassembling the sequences based on overlapping regions.3
Two primary methods of shotgun sequencing were developed: hierarchical shotgun sequencing (or clone-by-clone sequencing) and whole genome shotgun sequencing.4
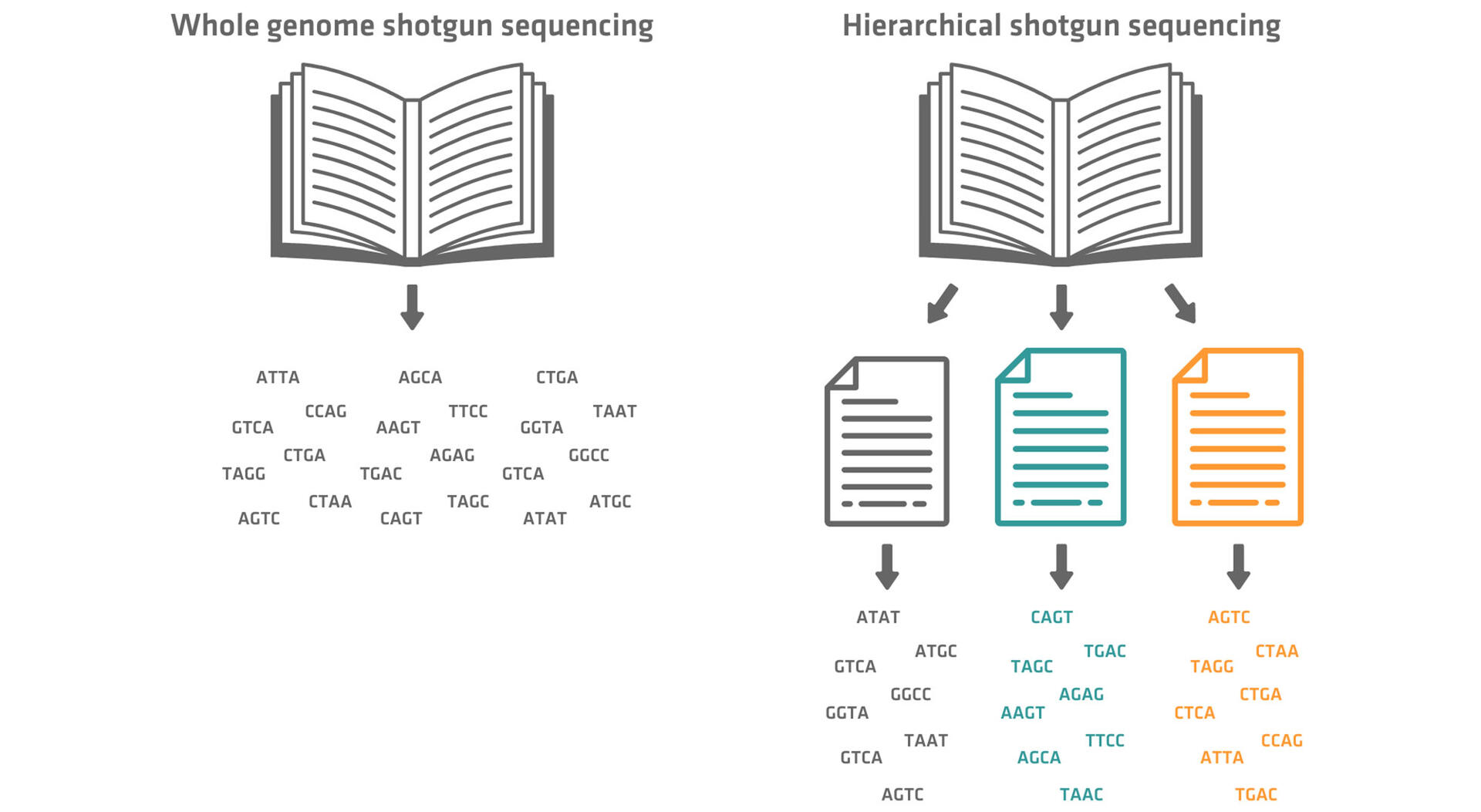
We will look at these methods in more detail below. Using a book analogy, this is how they compare:5
- Whole genome shotgun sequencing: Imagine a shredded book that needs to be reassembled.
- Hierarchical shotgun sequencing: Imagine pages torn out of a book. You reassemble the book from the pages. Then, each page is individually shredded and you need to reassemble these.
The Human Genome Project, which mapped our DNA for the first time, used hierarchical shotgun sequencing.6
How hierarchical shotgun sequencing works
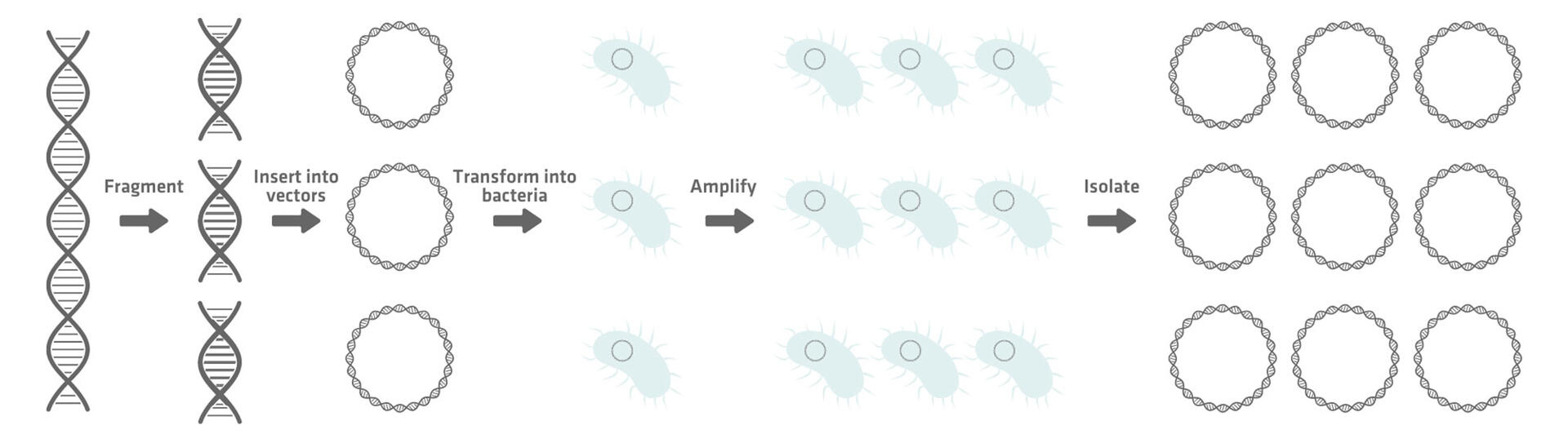
Hierarchical shotgun sequencing begins with randomly splitting the genome into sequences of 150-350 kbp, which are then inserted into vectors to create bacterial artificial chromosomes (BACs). These vectors are transformed into host bacterial cells, such as E. coli. When the bacterial cells divide, the DNA sequences are amplified and can subsequently be isolated from the host cells.5
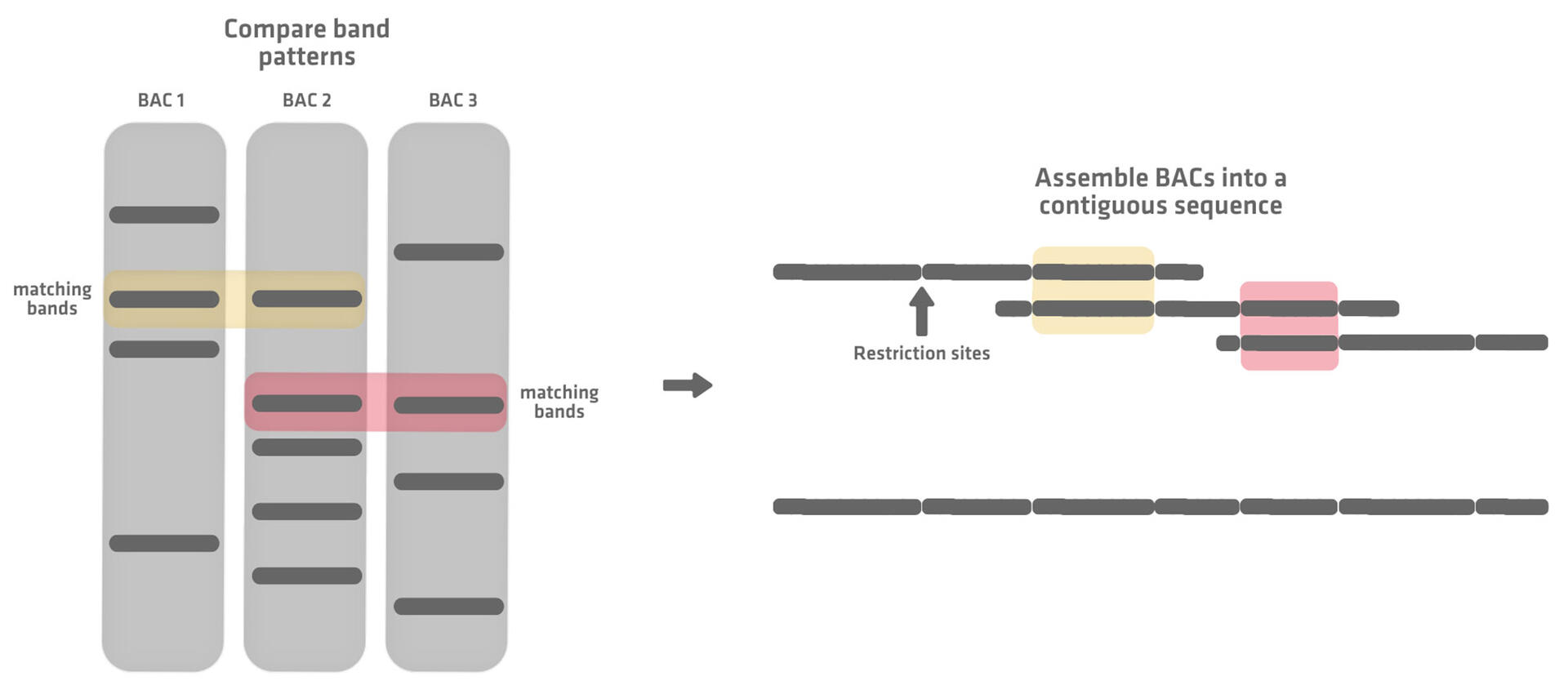
The next step is to create a genomic map or scaffold by reassembling the individual BACs. This is done by adding restriction enzymes to each BAC. Restriction enzymes recognize short specific nucleotide sequences and cut the DNA strand at or near these restriction sites, fragmenting each BAC into DNA sequences that differ in number and length.
By running a gel for each fragmented BAC, unique band patterns are obtained. Comparing these allows you to identify overlapping BACs based on matching bands. The BACs can then be assembled into a contiguous sequence, also referred to as a BAC contig.5
As multiple copies of the original genome run through this workflow, a highly redundant BAC library is generated. Sequencing all the BACs would mean sequencing most of the genome several times, which is why only a subset of BACs with minimal overlap is selected for the subsequent sequencing step.5
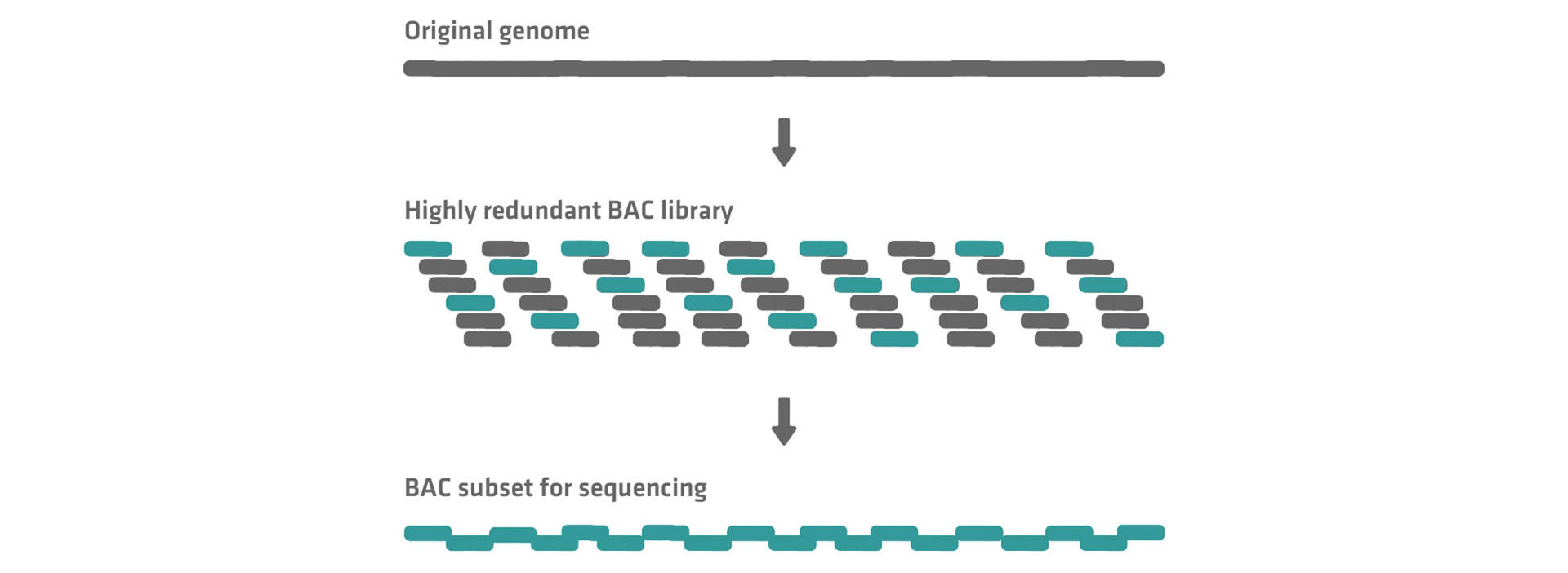
This subset of BACs is also called the minimum tiling path and is fragmented into shorter pieces that are sequenced and reassembled based on overlapping nucleotide sections to determine the original genome.5
How whole genome shotgun sequencing works
In contrast, whole genome shotgun sequencing simplifies the process by fragmenting the entire genome into short sequences, sequencing them in parallel, and then reassembling the reads based on overlapping nucleotide sequences.7
This approach, while seemingly straightforward, poses challenges, particularly with large genomes like the human genome. Reassembling millions of short reads is tricky and was even considered impossible when the method was first suggested, until mathematician Eugene Myers demonstrated that it is indeed feasible.7
Thanks to today's algorithms, whole genome shotgun sequencing has largely replaced hierarchical shotgun sequencing, and the preferred method for sequencing the individual DNA fragments is now Illumina short-read sequencing rather than Sanger sequencing. However, Illumina read lengths of 50-300 bp can push current reassembly algorithms to their limits, particularly for tasks such as de novo assembly or sequencing genomes with complex regions, structural variations and large stretches of repetitive sequences. To address these challenges, long-read techniques that generate fragments thousands of base pairs in length have been developed. For a detailed comparison of short- and long-read sequencing methods, please refer to this article: Short read vs long read sequencing.
Shotgun metagenomic sequencing
A recent extension of shotgun sequencing is shotgun metagenomic sequencing, used primarily in microbiome studies. Just like whole genome shotgun sequencing, its process involves fragmenting the sample DNA, sequencing the fragments, and using bioinformatics to stitch them back together. The difference is that the sample for shotgun metagenomic sequencing doesn't only contain DNA from one organism but from all the microorganisms present in a certain environment, for example, a soil or water sample in the case of environmental studies. This allows the identification of species and strains present in a sample, and for data to be obtained on the relative abundances of microorganisms and specific genes, such as antibiotic resistance genes in clinical samples.8
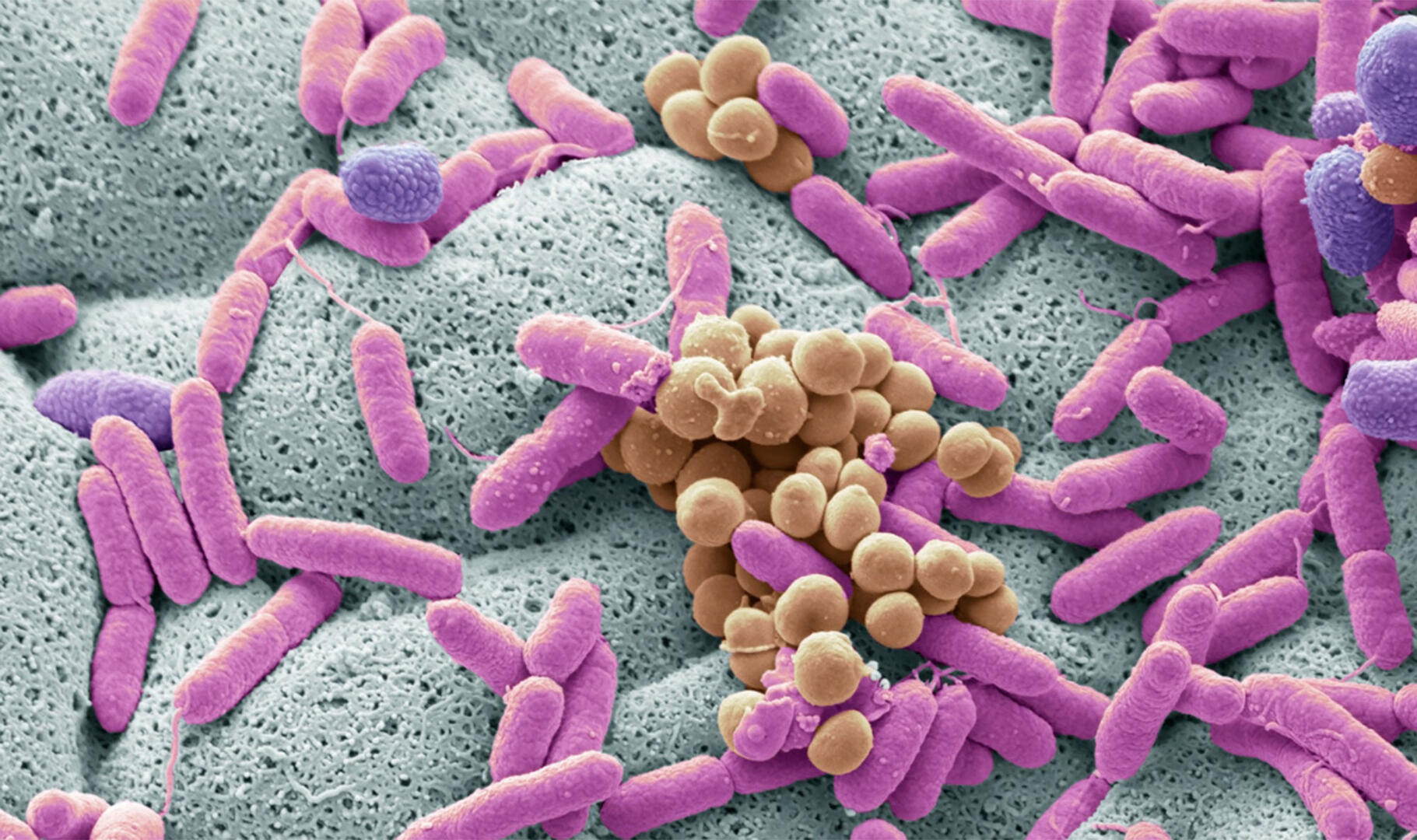
Shotgun metagenomic sequencing vs 16S rRNA gene sequencing
Traditionally, 16S rRNA gene sequencing has been the standard for microbiome studies. It targets a region of the 16S rRNA gene found in bacteria and archaea due to its hyper-variable regions between species. The sequencing process involves amplifying one or more hyper-variable regions of the 16S rRNA gene, sequencing the resulting amplicons and analyzing the data to identify and differentiate between microbial species.8 Choosing between shotgun metagenomic sequencing and 16S rRNA gene sequencing depends on your specific application, sample type, and budget. Below, we outline the pros and cons of both methods to help you make an informed decision.
Advantages of shotgun metagenomic sequencing
Shotgun metagenomic sequencing offers several advantages over 16S rRNA gene sequencing.
Firstly, it can identify all microorganisms present in a sample, whereas 16S rRNA gene sequencing is limited to identifying bacteria and archaea.
Secondly, by sequencing the entire genome of all microorganisms – the metagenome – shotgun metagenomic sequencing provides information on the functional potential of genes. It can, for example, determine whether bacterial cells have antibiotic resistance genes.
Lastly, 16S rRNA gene sequencing generally identifies bacteria only at the genus level, while shotgun metagenomic sequencing can identify bacteria and other microorganisms at the species level and sometimes even at the strain level.8
Disadvantages of shotgun metagenomic sequencing
Shotgun metagenomic sequencing, while powerful, also has some disadvantages compared to 16S rRNA gene sequencing.
A major drawback of shotgun metagenomic sequencing is its cost. Although it provides more comprehensive data, it is significantly more expensive than 16S rRNA gene sequencing. To address this, some researchers have started to use shallow shotgun sequencing. This method reduces sequencing depth (the number of times a certain nucleotide is sequenced) by combining more samples into a single sequencing run and uses a library preparation protocol with fewer reagents. This approach lowers costs while still providing data beyond the 16S rRNA gene region.8 Shallow shotgun sequencing has been shown to yield results similar to shotgun metagenomic sequencing for large-scale microbiome studies aimed at identifying species and functions. However, it cannot replace shotgun metagenomic sequencing for high resolution analyses.9
Another challenge with shotgun sequencing arises when dealing with samples containing a large amount of non-microbial DNA, such as human cheek swabs for oral microbiome studies. In these cases, reads from human DNA can obscure microbial reads, making analysis difficult. This can be avoided with 16S rRNA gene sequencing, which uses PCR to amplify only the microbial DNA regions of interest before sequencing.8
Furthermore, since 16S rRNA gene sequencing has been the traditional method for microbiome studies, there are extensive historical databases listing 16S rRNA gene sequences of microorganisms, helping to identify the ones present in a sample. In contrast, reference genomes for microbial species living in environments that have not been previously well characterized – for example, soil and marine samples – may be lacking. This means that shotgun metagenomic sequencing can sometimes provide less data than 16S rRNA gene sequencing. However, there are ongoing efforts to expand these databases with full microbial genomes.8 For example, Craig Venter and his team launched the Sorcerer II Global Ocean Sampling (GOS) Expedition in 2004, circumnavigating the globe for over 2 years.

They discovered millions of new genes and nearly 1000 genomes for uncultivated microbial lineages. Collaborative efforts and subsequent expeditions to various inland seas and lakes have further increased our understanding of microbial diversity in different water ecosystems, and, in 2017, additional expeditions even provided insights into microbes colonizing plastic pollution in marine environments.10
Conclusion
We hope that you now understand the history of shotgun sequencing and the journey from Sanger sequencing to modern NGS methods. This blog also outlined the different subtypes of shotgun sequencing, including hierarchical, whole genome and metagenomic shotgun sequencing, explaining how each method works. If you found this article informative and would like to stay updated with more blogs like this, please subscribe to our newsletter.
Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.