
What is genotyping and how does it work?
Written by Éva Mészáros
15. July 2022
Genotyping helps us discover unknown secrets in our DNA. It’s key in clinical research and diagnostics, and is even used in agriculture to tackle challenges such as climate change and hunger. The various genotyping methods are just as diverse as their applications so, in this article, we will provide an overview of this vast field, including definitions, the most important applications, and a review of the commonly used techniques.
Table of contents
What is genotyping?
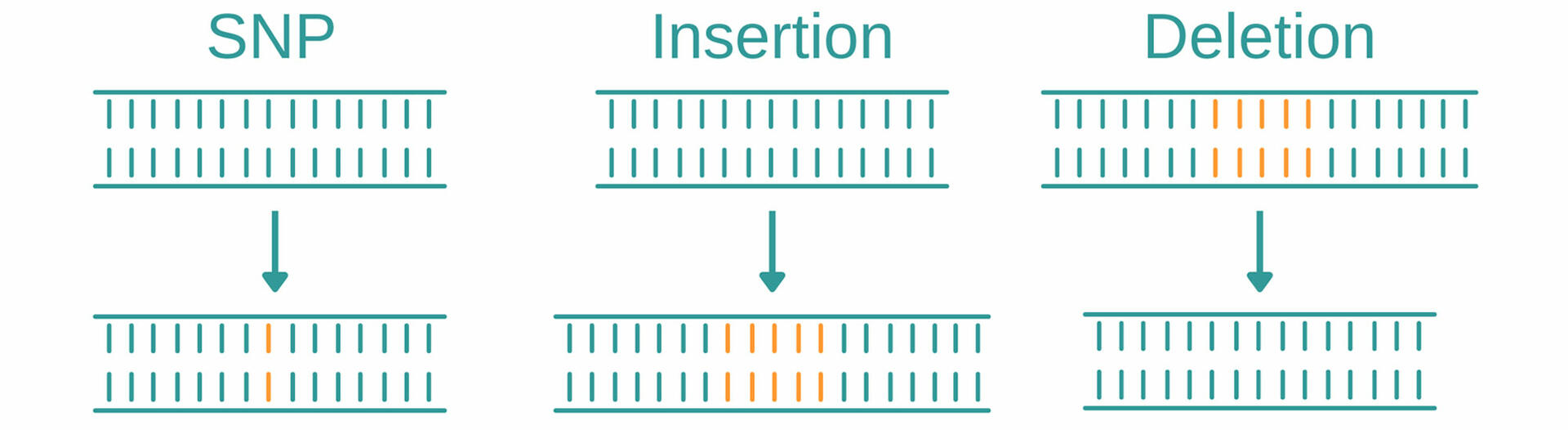
Genotyping is an umbrella term for several methods used to analyze variations in genomes between individual organisms. When comparing DNA sequences of an organism to a reference sequence or that of another sample, these methods look for variations such as single nucleotide polymorphisms, insertions, and deletions.
SNPs
Single nucleotide polymorphisms (abbreviated SNPs, pronounced 'snips') describe variations of a single base position in the DNA, and are the most common type of genetic mutation. In the human genome, for example, they occur on average about every 1000 nucleotides, which means that each human has 4 to 5 million SNPs in their DNA.1 Genotyping methods help to identify SNPs and determine if and how they impact factors such as health, disease, drug response, and other traits.
Insertions and deletions
Insertions and deletions, also called ‘indels’, are mutations that arise from the addition or loss of one or more nucleotides in a DNA segment. As with SNPs, insertions and deletions may have no (known) effect on an organism, but they could have negative effects, such as causing disease, or even be beneficial, for example, by making an organism resistant to certain pathogens.
What is genotyping used for?
Genotyping is used in a number of sectors, the most important ones being clinical research, clinical diagnostics and agriculture. Examples of how it can be used in these areas are given below.
Clinical research
If it can be statistically proven that individuals with a particular genomic variation are significantly more likely to be affected by a certain disease than the rest of the population, this variation potentially provides a marker of increased risk of developing that disease. Another major goal of performing disease-association studies is to create personalized drugs.
Clinical diagnostics
In diagnostics, genotyping has many different applications, including:
- Antimicrobial susceptibility testing: Genotyping can be used to determine whether a bacterial strain has specific resistance genes or genetic mutations that make it insusceptible to one or more antibiotic drugs.
- Genomic surveillance: Genotyping helps monitor viral variants during pandemics, helping to identify when a new variant becomes predominant, as with the SARS-CoV-2 virus.
- HLA typing: Genotyping techniques can be used to find donors and recipients with closely matched HLA (human leukocyte antigen) patterns, helping to reduce the risk of transplant rejection.
Agriculture
Genotyping can identify potential genes to improve crop and livestock breeding programs. This is of utmost importance if we consider that food production needs to increase dramatically in the next decades to feed the world's ever-growing population while adapting to and reducing climate change.2
How different genotyping methods work
Since genotyping is a broad area, we cannot explain all of the methods in this article. The following sections therefore focus on the most common techniques for detecting known SNPs and identifying new ones.
Genotyping by PCR
The two most common PCR methods to detect SNPs are amplification refractory mutation system PCR (ARMS PCR) and real-time PCR (qPCR).
ARMS PCR
For ARMS PCR, also called allele-specific PCR, you need to add four different primers to your master mix. The first primer pair is designed to amplify the DNA sequence containing the SNP of interest (red). The two other primers are sequence-specific for the forward strand of the wild type (Wt) allele (yellow) and the reverse strand of the mutant allele (orange):3
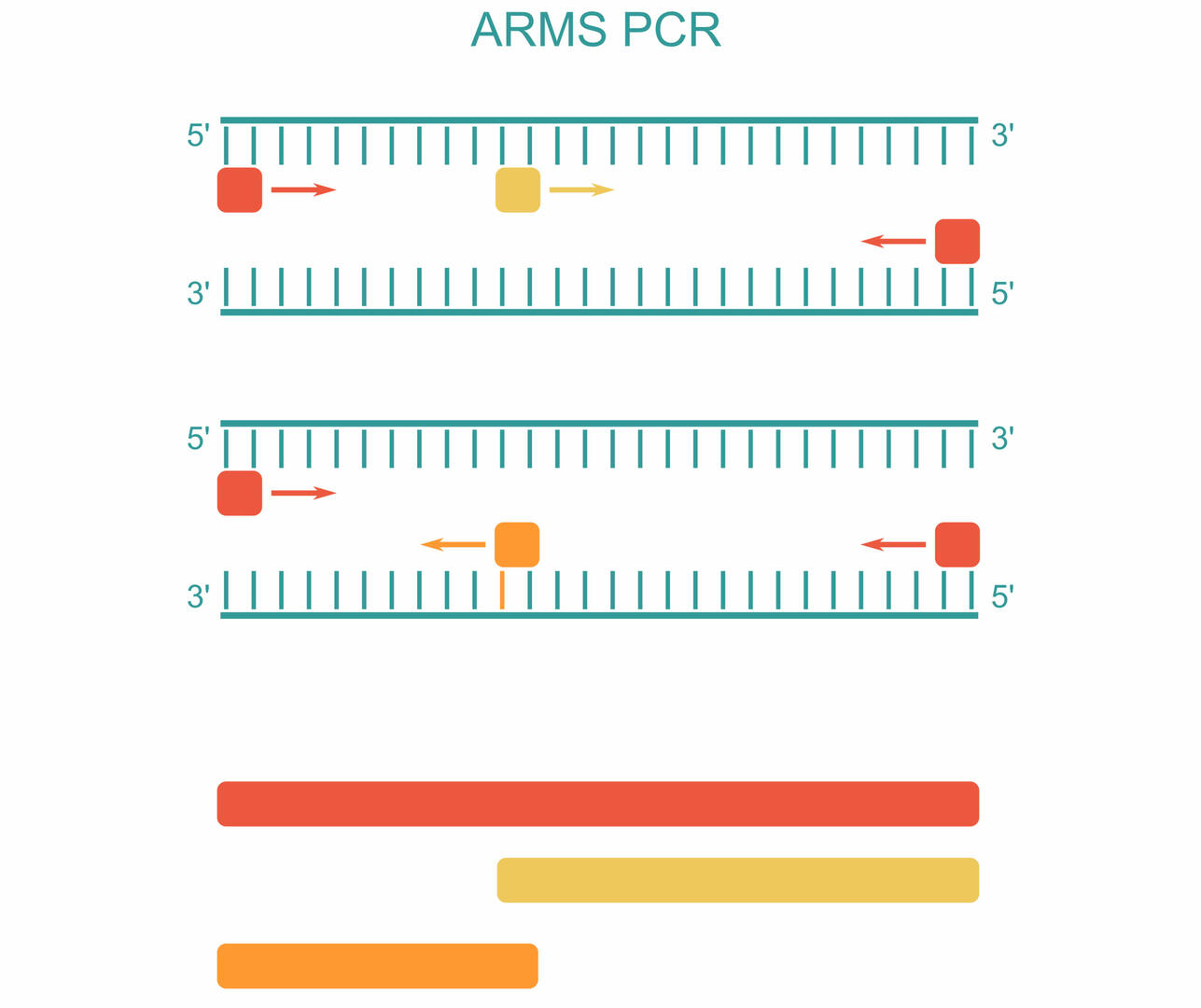
As you can see in the example above, three PCR products can be produced during amplification:3
- A long sequence produced by the first primer pair (red)
- A short sequence produced by the forward primer of the Wt allele and the reverse primer of the first primer pair (yellow)
- A short sequence produced by the reverse primer of the mutant allele and the forward primer of the first primer pair (orange)
To see what PCR products are present in your sample, you can run an agarose gel after amplification. If your sample is homozygous, you will only see two PCR products: the long sequence plus one of the short sequences, depending on whether the organism analyzed possesses two versions of the Wt allele or two versions of the mutant allele. If your sample is heterozygous, all three PCR products will be produced.3
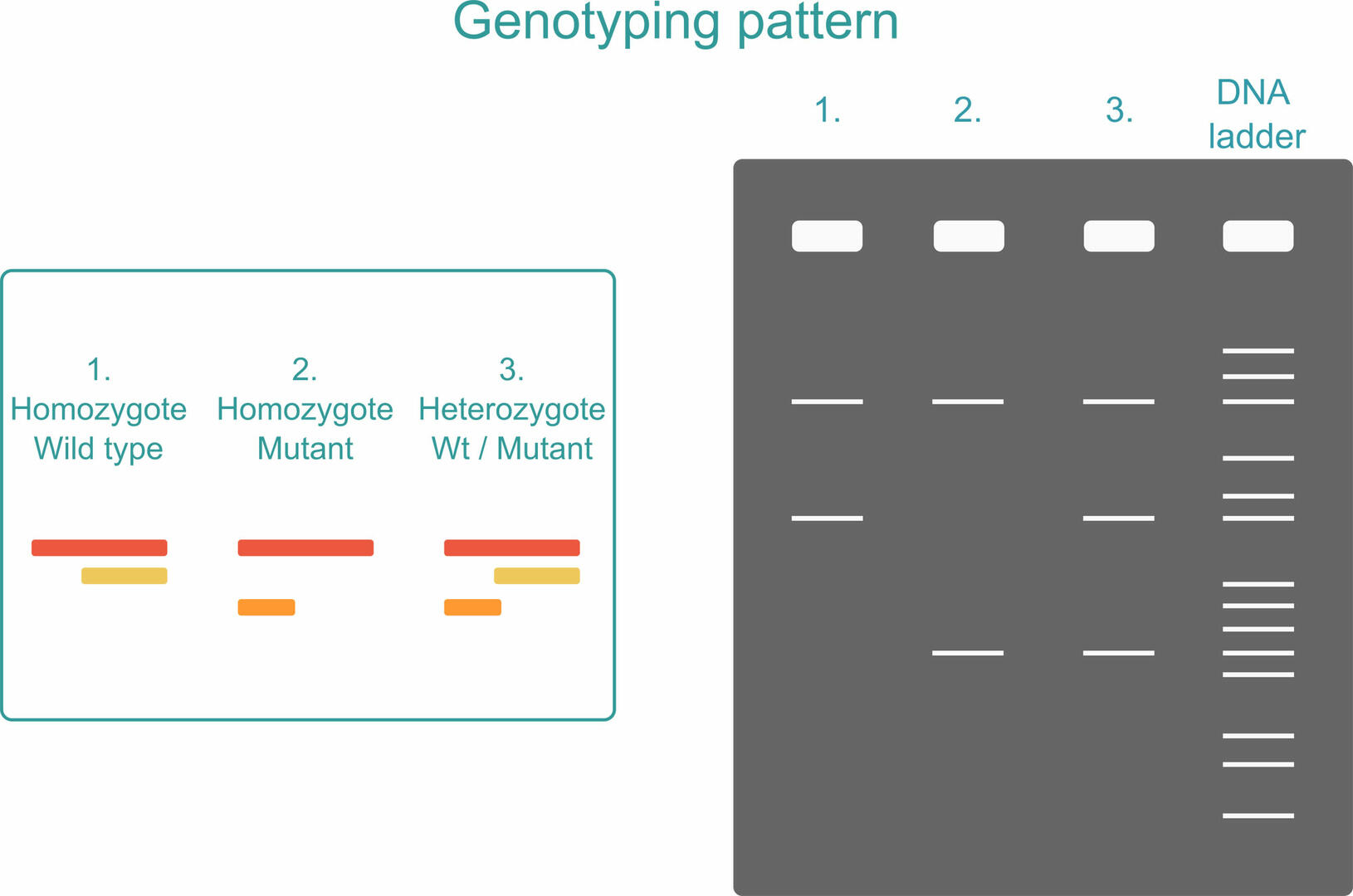
To ensure that you can distinguish the two short sequences from one another, the SNP should be located closer to either the forward or reverse primer of the first primer pair. It cannot be exactly in the middle as the two short PCR products would then be of equal length.3 You also have to reduce the number of thermal cycles when performing an ARMS PCR reaction. Only 22 to 25 cycles are recommended to reduce the risk of false-positive results. If the number of thermal cycles is increased, primers designed to anneal to the mutant allele could non-specifically amplify Wt alleles if their concentration is much higher. A second measure for reducing the risk of false-positive results is to include internal standards.4,5
ARMS PCR is mostly used in low throughput settings, because you can only detect one SNP at a time in one sample aliquot, and because running a gel for every PCR reaction is time consuming. It is, however, the method of choice for the diagnosis of the blood disorders thalassemia – which results in lower hemoglobin production – and sickle cell anemia, which causes unusually-shaped red blood cells. Its advantage compared to other methods – such as DNA microarrays and next generation sequencing, both described below – is that it doesn't rely on restriction digestion, which means that it's more accurate and can be used when restriction sites are absent in the DNA sequence of interest.4
qPCR
If you want to use qPCR to detect SNPs in a sample, slightly adapt the standard TaqMan qPCR workflow by adding two probes with different reporter dyes to the master mix. One probe needs to be designed to bind to the Wt allele, and the other one to the mutant allele of the SNP of interest. After amplification, you will either detect mostly one of the fluorescent signals if your sample is homozygous, or similar amounts of both signals if your sample is heterozygous. You will therefore see three clusters when plotting the signals obtained from several samples in a single allelic discrimination plot:6
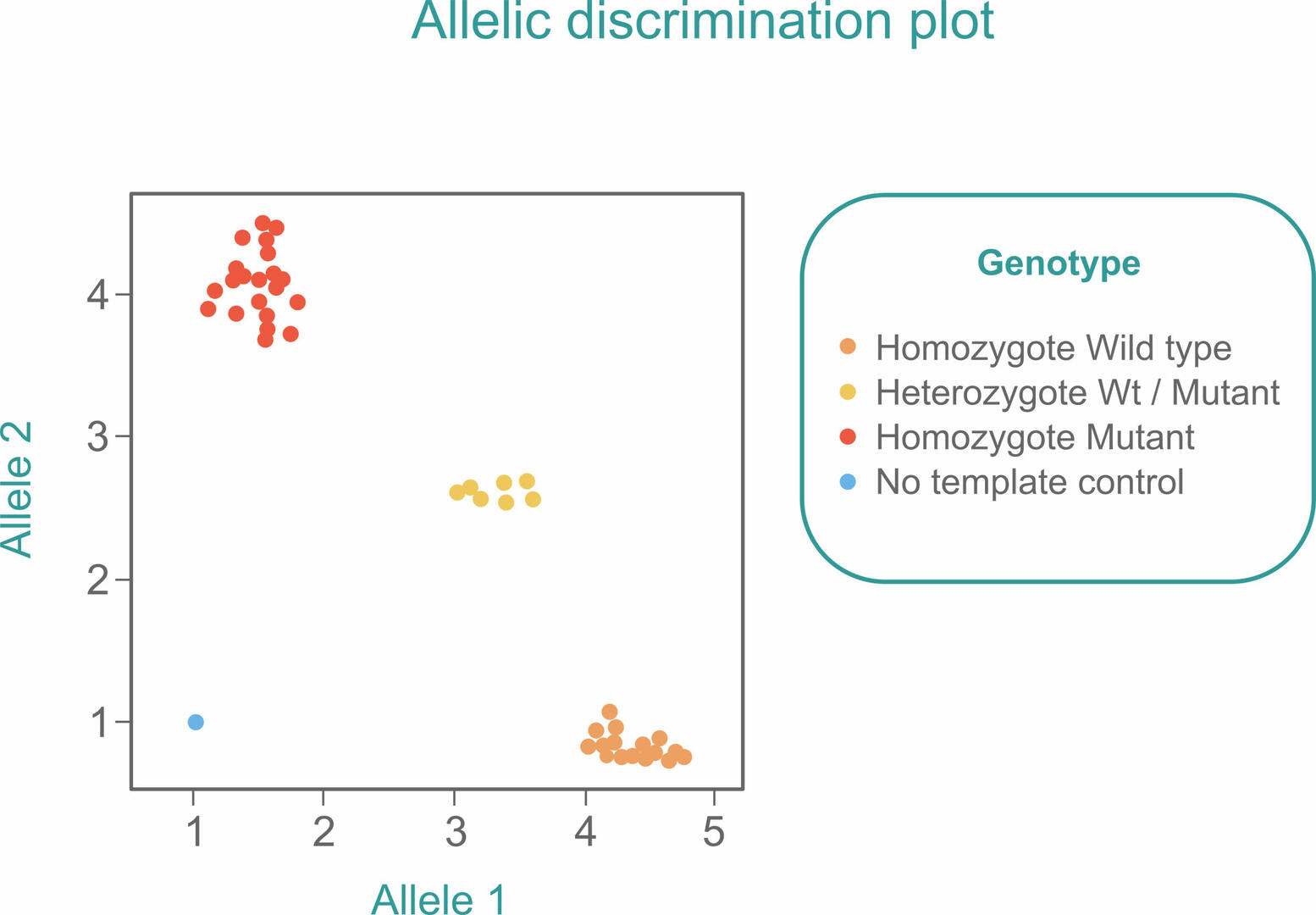
As in a standard qPCR reaction, you should also include a no template control (blue) to ensure that you would detect contamination early.
qPCR experiments can be set up in space-saving 96 or 384 well plates, making it an excellent method for workflows with large numbers of samples. However, as you can only detect one known SNP at a time in one sample aliquot, it is not suitable for analyzing more than a few SNPs.
DNA microarrays
A DNA microarray, sometimes called a DNA chip, is a collection of microscopic DNA spots attached to a solid surface. Each spot contains thousands of copies of a specific, single-stranded DNA sequence, known as probes.7,8,9,10 To detect SNPs using DNA microarray experiments, each probe needs to specifically bind to either the Wt or the mutant allele of a SNP of interest.
You then proceed as follows to determine the SNPs present in your sample:8,10
- Denature the DNA, cut the ssDNA into fragments, and label the fragments using a fluorescent dye.
- Insert the sample into the microarray, and allow the ssDNA fragments to hybridize to the probes.
- Wash away unbound DNA fragments, then scan and read the microarray. Fluorescent spots indicate the SNPs present in the sample.
You can also mix two samples – e.g., a patient sample labeled in red, and a control sample labeled in green – and add them to the microarray at the same time. Yellow spots indicate shared SNPs, whereas green and red spots represent SNPs that are only contained in the control or patient samples, respectively.8,10
As thousands of different probes can be attached to a single DNA microarray, this method is ideal for experiments where a large number of SNPs needs to be detected in a sample. It is, however, less suitable for high throughput settings, because you would need a lot of space and time to set up and analyze microarrays for hundreds of samples.
Integrated fluid circuits
For workflows with high sample numbers, or experiments where probes need to be added, removed or replaced on demand, integrated fluid circuits (IFCs) are preferred over DNA microarrays. Instead of using probes that are fixed or pre-spotted onto a solid surface, IFCs have sample and assay inlets:
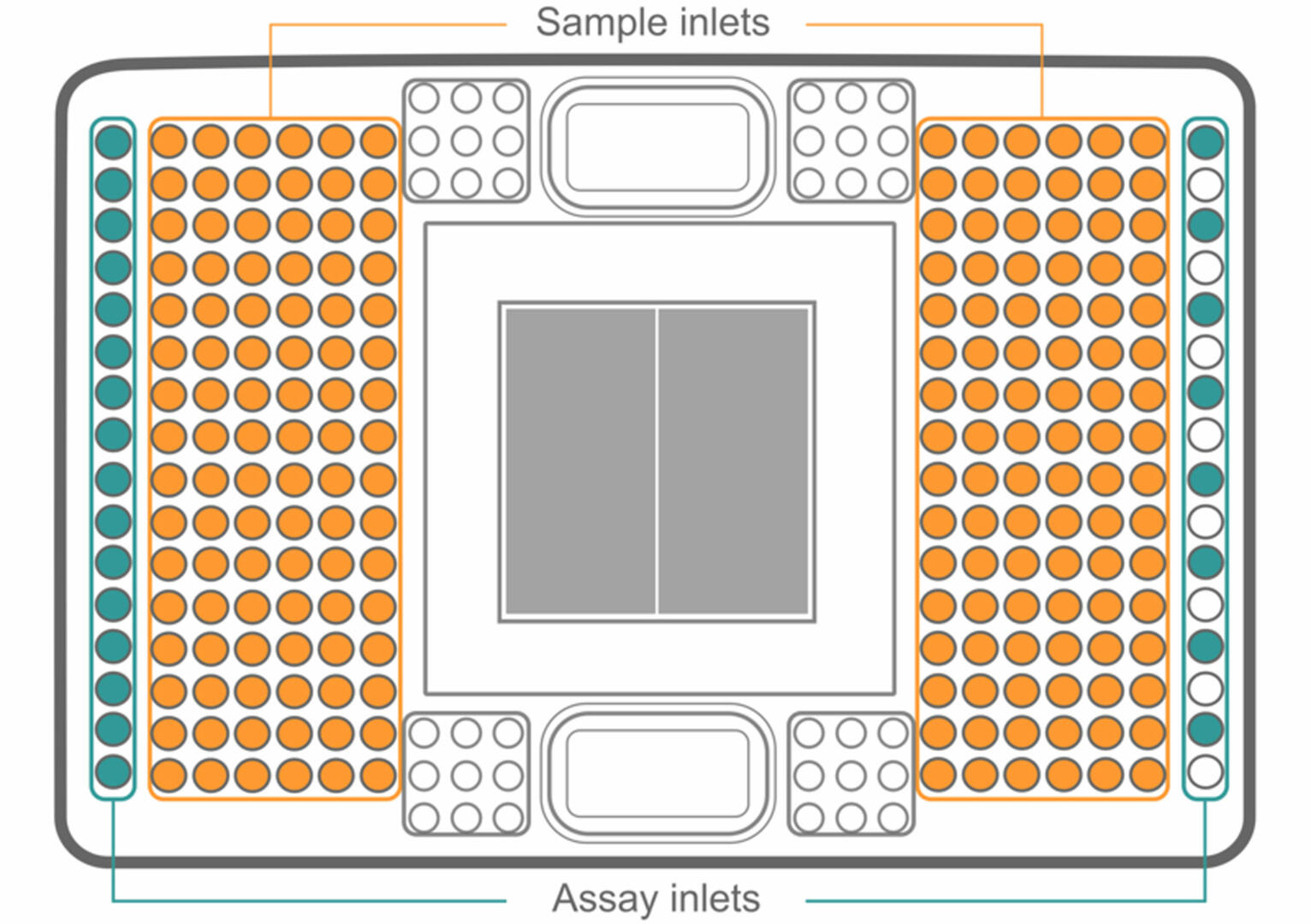
To set up an IFC, first load your samples into the plate. Then, add a different master mix to each assay inlet. As with genotyping by qPCR, every master mix has to contain a primer pair binding to a DNA sequence containing a SNP of interest, as well as probes specific to both the Wt and mutant allele of this SNP.
Once your IFC is set up, the samples and master mixes pass through a complex network of fluid lines, valves and membranes, so that each sample is combined with each master mix in a microfluidic reaction chamber (the grey square in the center). Using a 192.24 IFC (as shown in the image above), this results in 4608 different combinations. Using a special qPCR cycler11, you can then analyze your samples for SNPs as described above.
If you're using IFCs, you might want to automate the loading steps, as the manual pipetting of such a complex format can be error-prone and tedious. An application note of how an IFC can be loaded on the ASSIST PLUS pipetting robot can be found here.
MassARRAY SNP genotyping
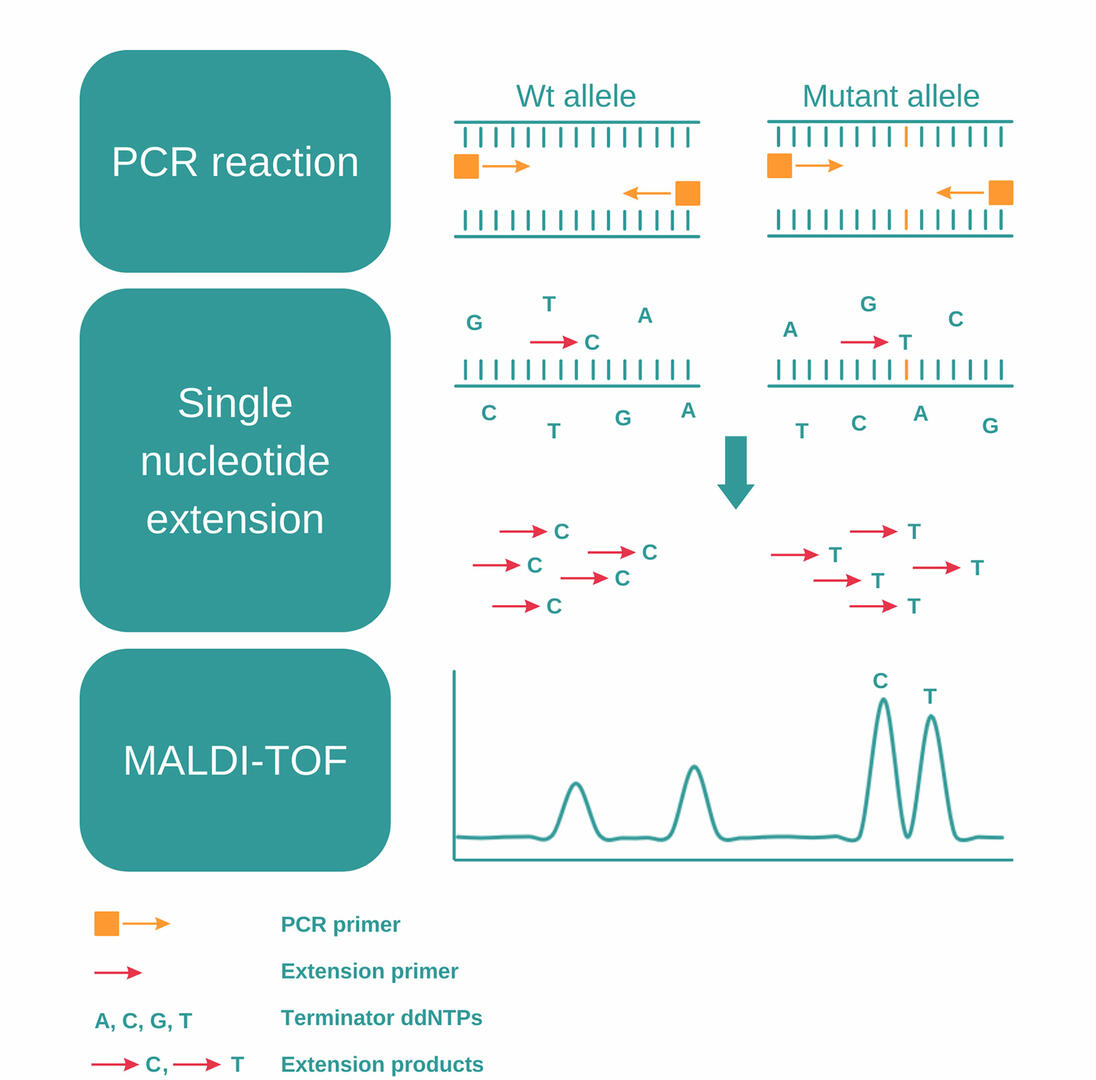
MassARRAY SNP genotyping uses a combination of PCR, single nucleotide extension and MALDI-TOF to detect SNPs, and consists of the following steps:12,13
- Perform a PCR reaction to amplify a DNA sequence containing a SNP of interest.
- Eliminate unincorporated nucleotides using shrimp alkaline phosphatase.
- Add extension primers designed to anneal just next to the SNP, together with extension enzymes and terminator ddNTPs (dideoxynucleotides).
- During the second PCR reaction, the primers will be extended by a single dideoxynucleotide, matching the nucleotide of the SNP.
- As the four terminator ddNTPs – A, C, G, and T – have small differences in molecular mass, you can then analyze these PCR products and detect SNPs using MALDI-TOF.
MALDI-TOF
MALDI-TOF is a two-phase mass spectrometry procedure. In the initial step, samples fixed in a crystalline matrix are vaporized and ionized by a laser. High voltage is then applied to accelerate the charged particles, and a detector measures the time of flight of each particle, enabling it to determine their molecular masses.
MassARRAY SNP genotyping is ideal for high throughput labs. It allows you to process up to 384 samples in parallel and, by using a multiplex approach, you can increase the number of SNPs detected in a single sample aliquot to dozens.
Genotyping by sequencing
The methods described above can only be used for the detection of known SNPs, as you need to be able to design primers and probes for the DNA sequence containing the SNP of interest. In contrast, genotyping by sequencing (GBS) – also called next generation genotyping – can be used for the identification of new SNPs.
The most commonly used DNA sequencing method in genotyping is next generation sequencing.
Next generation sequencing
Next generation sequencing (NGS) is a technique used to sequence millions of DNA fragments in parallel. First, a library is prepared by fragmentation, purification and amplification of the DNA sample. Individual fragments are then physically isolated by attachment to solid surfaces or small beads. The sequence of each of these fragments is determined simultaneously, and computationally aligned against a ‘normal reference’ genome. This enables the detection of many sequence alterations, such as SNPs, in a single reaction.
GBS is the only method capable of identifying unknown SNPs. It can be used to determine a large number of SNPs in a small number of samples, as millions of sequences can be analyzed in parallel, but you have to set up a separate experiment for every sample.
Conclusion
While the methods described above have helped to discover countless genetic variants and their impact, we're still far from understanding what every piece of our DNA does. Genotyping will likely play a major role in unlocking these secrets, and could ultimately improve our wellbeing by advancing personalized medicine, translating genetic mutations into healthy crops and livestock to feed future generations, and much more.
Did you like this article? Subscribe to our newsletter for more blog posts like this!



