Single-cell RNA sequencing methods: plate-, droplet- and microwell-based approaches
Written by Stefan Zeyen · 17. October 2025
Bulk and single-cell RNA sequencing (scRNA-seq) emerged almost simultaneously, with the first bulk RNA-seq study published in 2008, and the first single-cell publication following just a year later.1,2 While bulk sequencing quickly became the dominant approach, scRNA-seq has gained traction over the past decade. In 2015, only 6 % of RNA-seq publications used single-cell methods; today, the proportion has risen to more than a third,3 and a diverse set of techniques has been developed to capture and analyze single-cell transcriptomes.
In this blog post, we’ll walk through the major categories of scRNA-seq methods – plate-, droplet- and microwell-based – and explain how each one works. If you're looking for a more general introduction to single-cell sequencing, please refer to our blog 'Single-cell sequencing: an introduction to decoding cellular diversity'.
Table of contents
Plate-based methods
The first scRNA-seq methods used plate-based formats, in which individual cells were sorted into separate wells for processing.
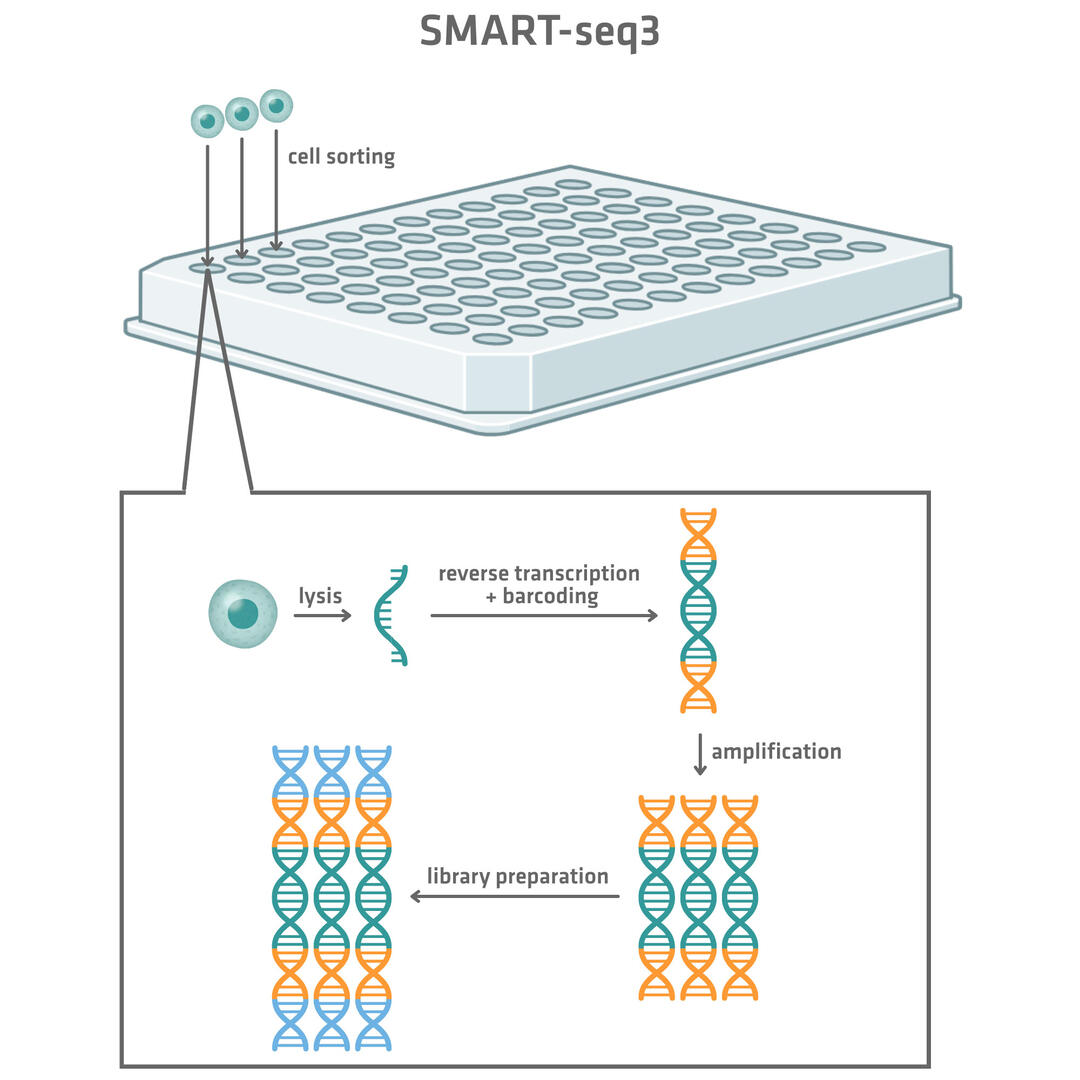
SMART-seq (switching mechanism at the 5' end of RNA template sequencing) is an example of an early plate-based sequencing method (Figure 1). It uses a flow cytometer to sort single cells into wells of 96 or 384 well plates. Once sorted, the cells are lysed and the RNA sequences are reverse transcribed, amplified and prepared into sequencing-ready libraries. The original SMART-seq protocols did not include unique molecular barcodes that would allow the assignment of sequencing reads to individual cells. As a result, libraries from each cell had to be prepared and sequenced separately. Over time, the method has evolved, and the SMART-seq3 now incorporates cell-specific barcodes during library preparation. This advancement allows the pooling, combined processing and simultaneous sequencing of multiple cells, streamlining the workflow and increasing throughput.4
CEL-seq (cell expression by linear amplification and sequencing) is a plate-based method that enables the pooling and combined sequencing of RNA sequences from several cells right from the beginning. Individual cells are sorted into separate microwells and lysed. However, a unique barcoded primer is always used for reverse transcription of the RNA into cDNA, allowing the cDNA from all the wells to be pooled prior to amplification and sequencing platform-specific adapter ligation.
More modern plate-based scRNA-seq methods use combinatorial indexing to barcode the genetic material of the individual cells (Figure 2). These methods tag each cell with a longer combinatorial barcode composed of several shorter barcodes. Here's how the technique works:
- Fixed, permeabilized cells are created that allow reagents to flow in and out. These can be used as reaction compartments in the following steps.
- These cells are sorted into wells of a 96, 384 or 1,536 well plate. Several cells can be distributed into each well.
- The RNA of each cell is reverse transcribed and barcoded with a well-specific barcode.
- All the cells are pooled, mixed and redistributed into another 96, 384 or 1,536 well plate.
- In the second plate, the cDNA of each cell is tagged with a second well-specific barcode. This combination of barcodes allows sequencing reads to be assigned to single cells.
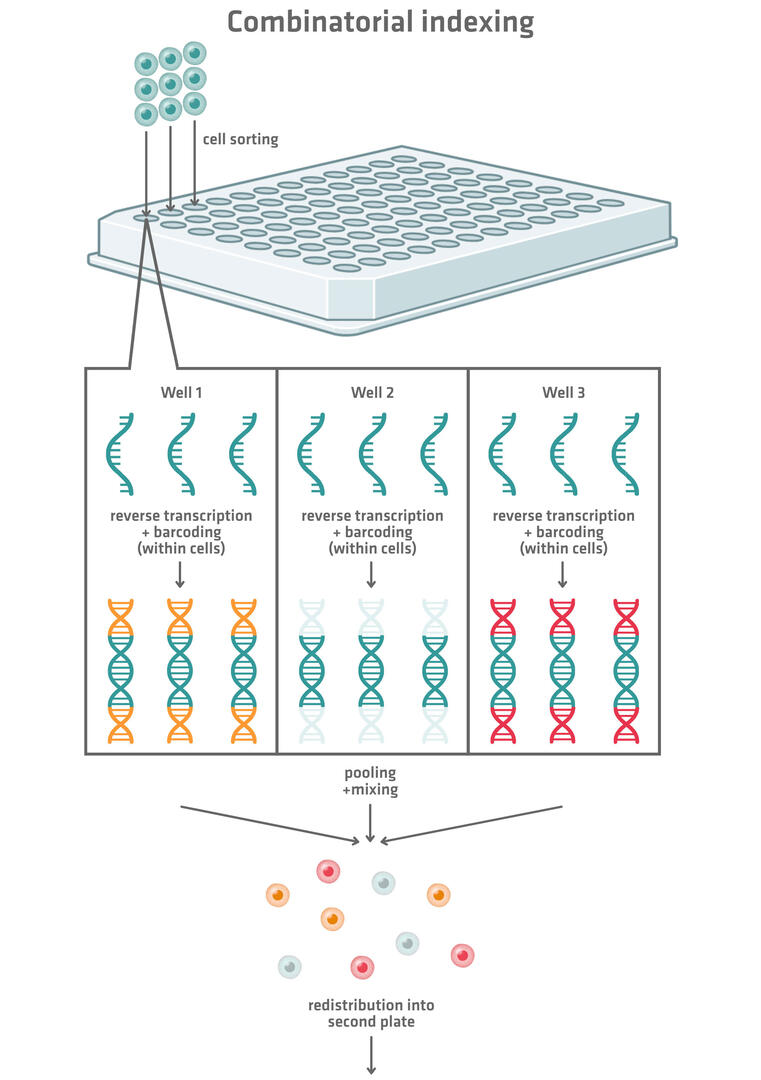
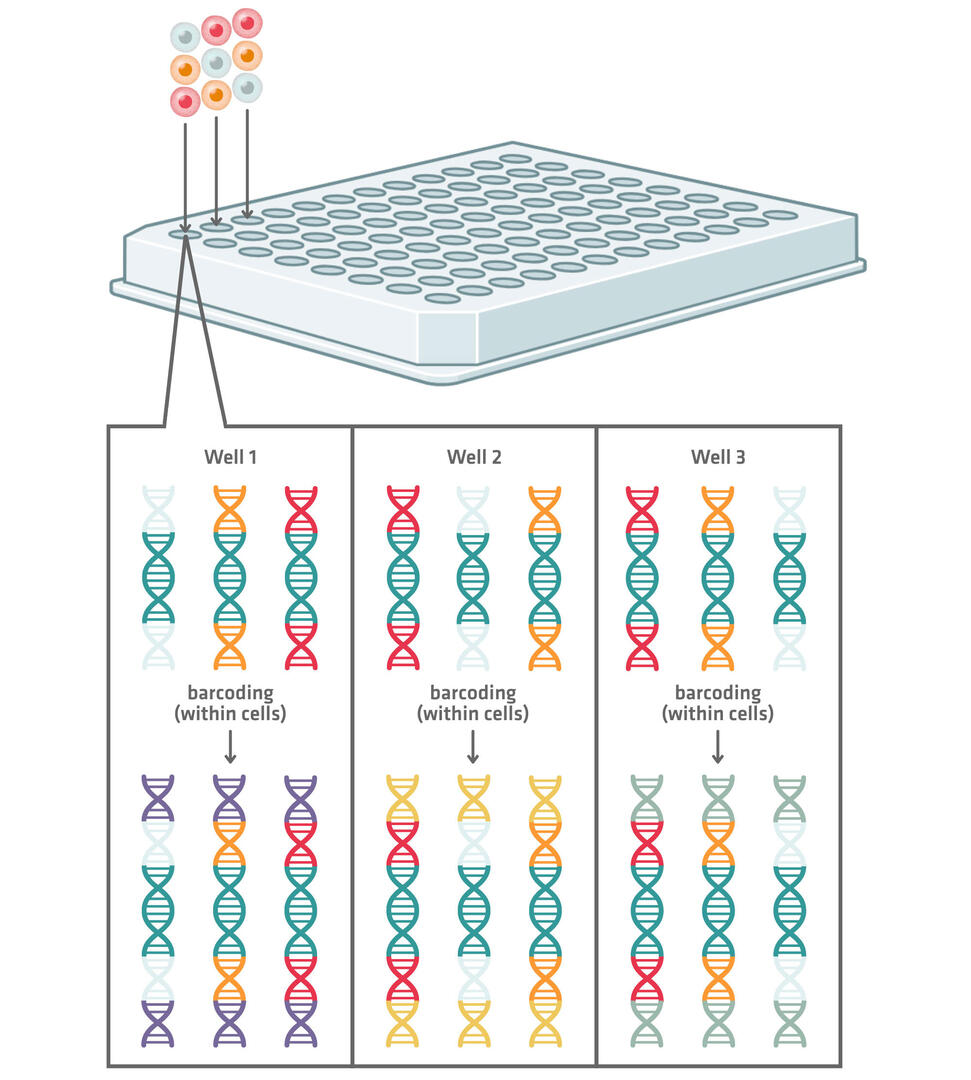
The choice of 96, 384 or 1,536 well plates for this method depends on the number of cells to be sequenced. Working with 96 well plates will result in about 10,000 possible well combinations. The chances that 2 cells end up in the same well in both plates should therefore be low enough if you are planning to sequence about 1,000 cells. Increasing the number of wells that you're working with increases the number of possible combinations, consequently enabling the barcoding of more cells while still keeping the risk of 2 cells receiving the same barcode combination low. For example, when working with 1,536 well plates, there are about 2.4 million different combinations. If this still isn't good enough, you could even add a third and fourth round of barcoding.
An example of a manufacturer offering combinatorial indexing for single-cell sequencing is Parse Biosciences. Its Evercode™ technology allows the processing of up to 1 million cells and 96 biological samples in parallel, and can be seamlessly integrated with our ASSIST PLUS pipetting robot
While SMART-seq3 and CEL-seq provide limited throughput, combinatorial indexing strategies significantly increase scalability. However, the microfluidic droplet- and microwell-based techniques described below are generally preferable for high throughput studies, as their greater miniaturization results in a lower sequencing cost per cell. Despite this, plate-based methods continue to be widely used due to their increased sensitivity.
Droplet-based methods
Droplet-based single-cell sequencing methods make use of microfluidics systems (Figure 3). We'll have a closer look at the techniques from DropSeq and 10x Genomics Chromium in more detail in this article.4
Both methods begin by mixing cells with beads featuring unique barcodes. This aqueous suspension is then combined with oil to create an emulsion of thousands of nanoliter-sized droplets, each ideally containing 1 cell and 1 bead. When the cells are lysed, their RNA molecules are released and hybridize to the barcoded bead within each droplet. In the DropSeq technique, the emulsion is then broken down and all the RNA molecules attached to the beads are reverse transcribed. In comparison, in the 10x Genomics Chromium system, beads are reversed transcribed before the emulsion is broken down. In both methods, the resulting cDNA is amplified and converted into sequencing-ready libraries. After sequencing, the bead-derived barcodes enable the assignment of reads back to individual cells.
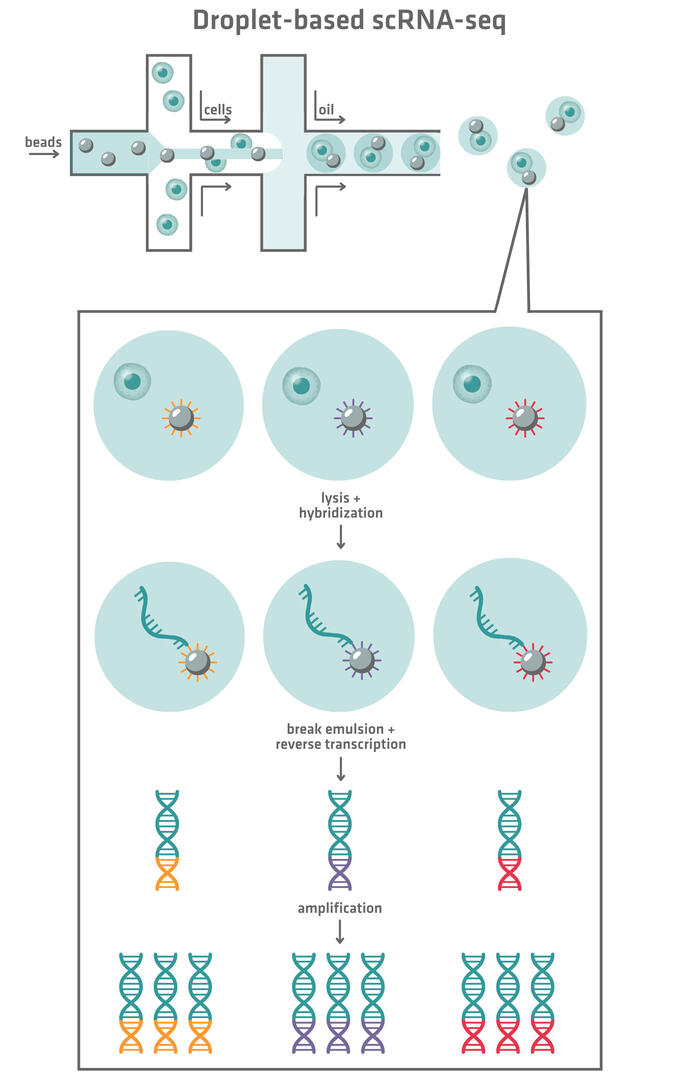
Ideally, the emulsion created by the microfluidics systems should only contain droplets with exactly 1 cell and 1 bead. However, in reality, there will be droplets that are completely empty, droplets that contain only a cell or bead, and droplets that contain 2 or more cells or beads. Empty droplets or droplets containing only 1 bead won't have any impact on your sequencing data quality, whereas droplets containing only cells will lower your sequencing yield. Droplets containing a cell and several beads, or several cells and a bead, are the most problematic. If the RNA molecules of 1 cell hybridize to several beads, the reads from 1 cell may be mistakenly assigned to several cells. On the other hand, if the RNA molecules of several cells hybridize to 1 bead, the reads from several cells may be assigned to a single cell. This is why droplets with more than 1 cell or bead need to be avoided.
In the DropSeq microfluidics system – the first droplet-based technique invented – the distribution of beads and cells is random. This means that both cells and beads need to be loaded at low concentrations to avoid the formation of too many droplets with 1 or more cells or beads.
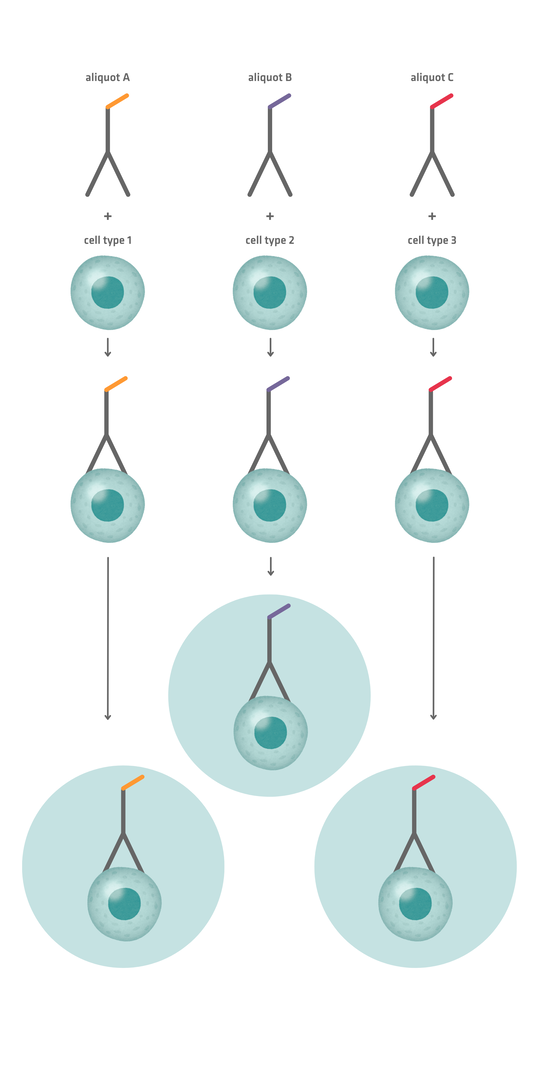
10x Genomics Chromium – which was released later – was engineered so that most droplets contain exactly 1 bead, increasing efficiency and enabling beads to be loaded at higher concentrations. However, cells still need to be loaded at low concentrations to avoid 2 cells being enclosed in a single droplet, unless computational methods are applied to identify and remove doublets (microwells containing more than 1 cell) from the sequencing data. One such strategy involves antibody labeling: by binding an antibody against a universal surface antigen and labeling different aliquots of this antibody with distinct barcodes, then mixing each aliquot with a sample, each sample can be uniquely labeled before pooling. When pooled samples are added to the microfluidics system, doublets are likely to consist of cells from different samples. They should therefore feature different antibody barcodes, allowing you to exclude these sequencing reads from data analysis.
Is your lab using or planning to use the 10x Genomics Chromium method? Check out our application note about how the ASSIST PLUS can be used to streamline the Flex Apex library preparation workflow.
Microwell-based methods
A third single-cell sequencing approach is to use microwells instead of droplets to isolate individual cells. Microwell plate systems require loading a chip containing hundreds of thousands of tiny wells with uniquely labeled beads. As the beads settle, each one typically occupies an individual well. The chip can then be loaded with cells, which also ideally settle in individual wells. The subsequent process is very similar to droplet-based methods: cells are lysed, RNA molecules hybridize to the bead and are reverse transcribed, and the resulting cDNA is amplified and prepared for sequencing. During sequencing, all the cDNA fragments can be pooled, because the uniquely labeled beads will have tagged the genetic material from each cell with a different barcode.
Note that doublets can also be a problem during microwell-based methods. You will need to apply the same computational techniques used for droplet-based methods to remove these from your sequencing data if you want to increase the number of cells loaded per chip.
The key distinction between droplet- and microwell-based methods lies in how cells are isolated: either encapsulated in droplets or captured in wells on a chip. For microwell platforms, specialized chips can increase sequencing costs per cell, and the chip size can limit throughput, resulting in slightly lower sample volumes compared to droplet-based systems. However, these chips provide greater control over cell capture, and are therefore better suited for low volume and precious samples. On the other hand, droplet-based techniques require substantial upfront investment in microfluidics equipment, making them less cost effective for smaller studies.
Conclusion
From plate-based protocols to the development of droplet and microwell systems, scRNA-seq methods have steadily expanded the possibilities for studying gene expression at the cellular level. Each approach relies on different principles of isolating and barcoding single cells, but all share the same goal of enabling researchers to capture transcriptomes with single-cell resolution. The pros and cons of each method are summarized in the table below.
| Plate-based scRNA-seq | Droplet-based scRNA-seq | Microwell-based scRNA-seq | |
| Throughput | Lowest, although combinatorial indexing improves scalability | Highest | Intermediate |
| Cost per cell | Highest, due to greater reagent consumption | Lowest, due to microfluidics miniaturization | Intermediate |
| Sensitivity | Highest | Lower than plate-based | Lower than plate-based |
| Workflow | Flexible but labor intensive (involves manual cell sorting and numerous pipetting steps) | Highly automated, but requires expensive microfluidics equipment | Partially automated |
| Best for | Smaller scale, in-depth studies | Large-scale studies | Medium- to large-scale studies |
Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.
About the author