Short read vs long read sequencing
Written by Éva Mészáros · 22. April 2024

The landscape of DNA sequencing is continuously advancing, with new players and techniques to decode genetic information emerging. Today, two main next generation sequencing (NGS) methods are available: short read and long read sequencing. Each approach has its own strengths and weaknesses, and comprises an array of technologies that can be further classified into subcategories.
This article is here to guide you through how these different sequencing technologies operate, and to highlight the key advantages and drawbacks of both short read and long read sequencing. Our aim is to provide you with a clear understanding, helping you to decide which method is best suited to your application.
Table of contents
Short read sequencing
Short read sequencing, with read lengths usually ranging from 50 to 300 base pairs, is the most widely used approach. It covers a range of techniques, each employing unique methods to decipher genetic information. Broadly, the platforms available fall into three categories: sequencing by synthesis (SBS), sequencing by binding (SBB), and sequencing by ligation (SBL).
Sequencing by synthesis
SBS platforms make use of polymerase enzymes that replicate single stranded DNA fragments to determine the nucleotide sequence of a sample. Two primary methods are available to detect what type of nucleotide is incorporated at a certain location.
The first involves the use of fluorescently-labeled nucleotides paired with reversible blockers that prevent additional nucleotides from attaching. Once a labeled nucleotide is incorporated by the polymerase, the reaction halts, allowing the identification of the newly added base. The fluorescent label and reversible blocker are then chemically removed to enable the addition of the next nucleotide.
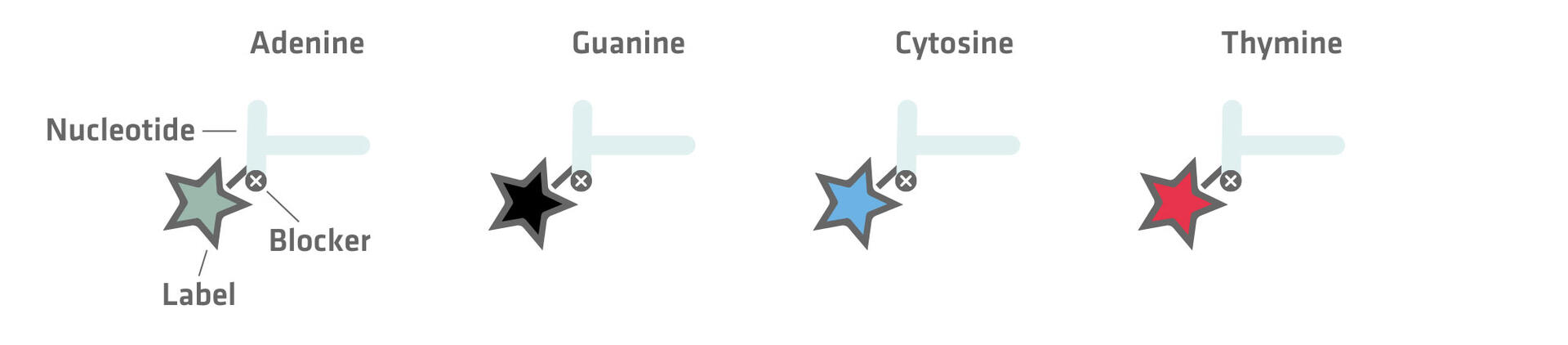
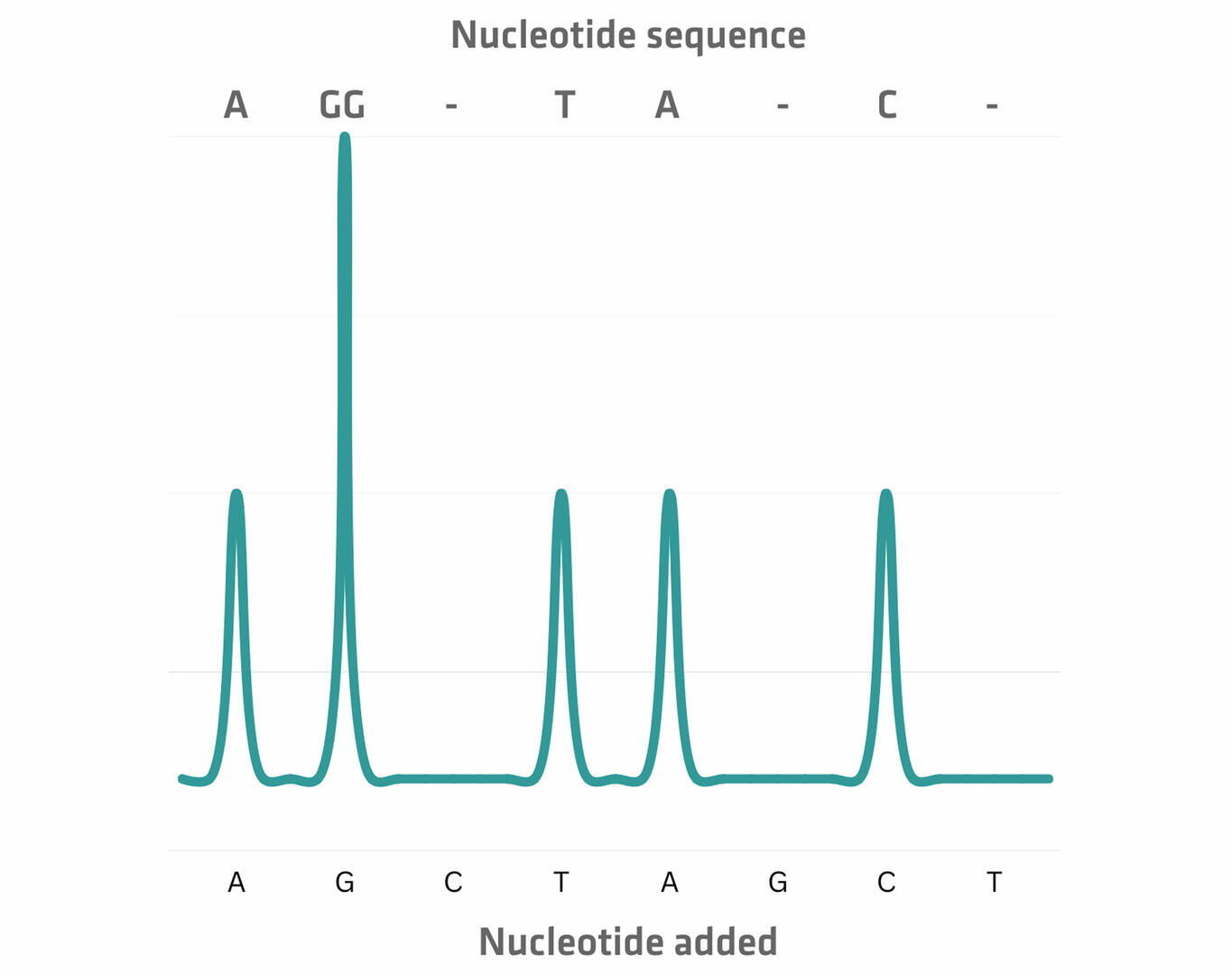
The second method uses unmodified nucleotides that are introduced sequentially. Let's suppose that the first nucleotide added is adenine. Its incorporation triggers the release of hydrogen ions and pyrophosphate, which the sequencing equipment can detect as a signal that is proportional to the number of nucleotides added. In the absence of incorporation, no signal would be emitted. Following each detection step, the unincorporated nucleotides are washed away, and the next type of nucleotide is added.
Examples of SBS methods, including links to a more in-depth explanation of how they work, can be found here:
Sequencing by binding
SBB platforms also use polymerase enzymes to replicate single stranded DNA fragments. However, the process of extending the sample strand by one nucleotide is split into two distinct steps.
At the beginning of an SBB workflow, you have a single stranded DNA fragment with a primer and a reversible blocker attached to it (step 1 in the image below). Fluorescently-labeled nucleotides are then introduced, the nucleotide type complementary to the template strand binds to it, and the sequencing platform registers its fluorescent signal (step 2 in the image below). However, due to the blocker attached to the preceding base, the labeled nucleotide cannot be incorporated by the polymerase, and is washed away (step 3 in the image below). Following the washing step, the blocker on the previous base is chemically removed, and unlabeled nucleotides carrying reversible blockers are introduced. This allows the polymerase to extend the DNA strand by a single base (step 4 in the image below).
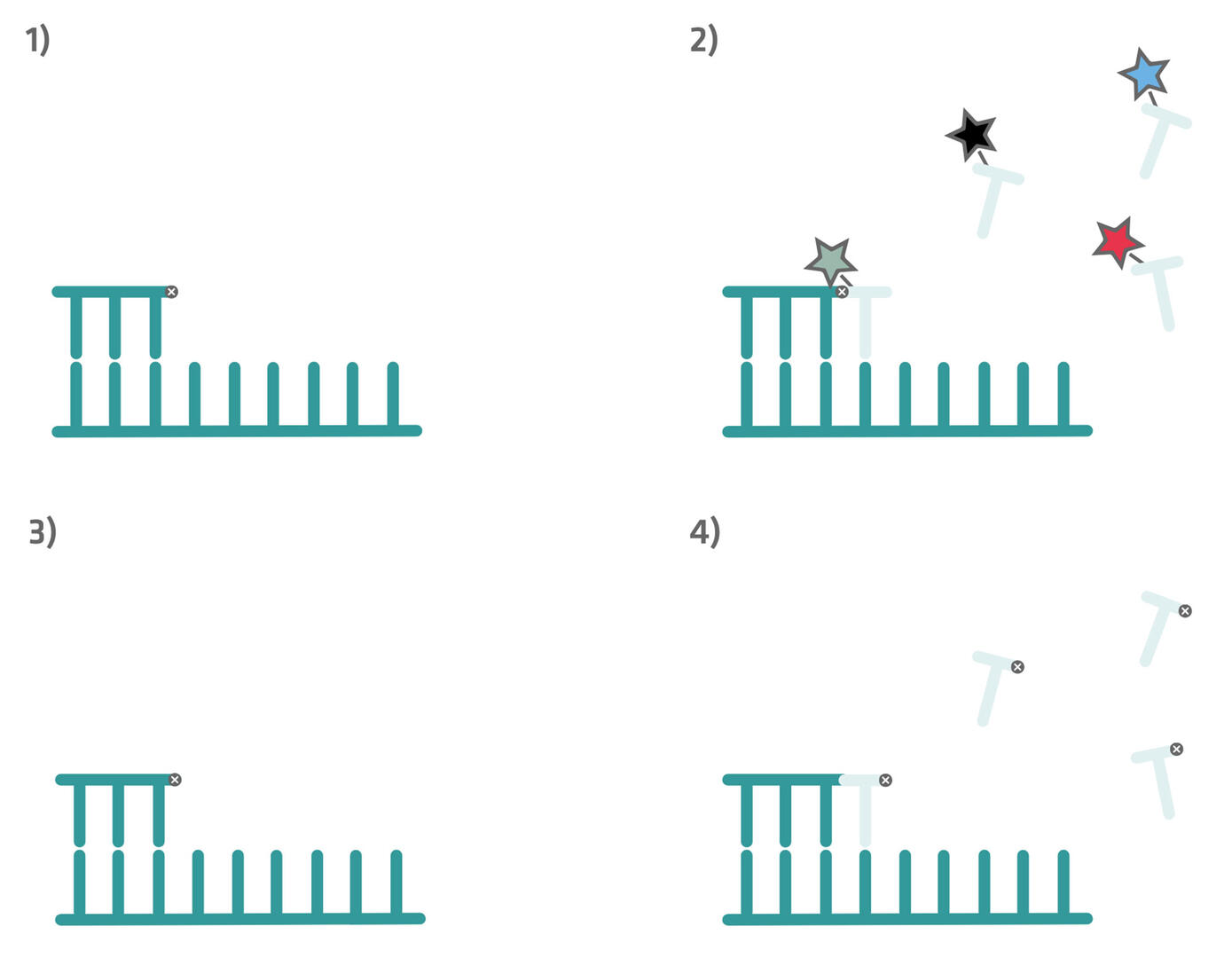
The cycle of binding, signal detection, washing, and extension is repeated iteratively, until the entire DNA fragment has been sequenced.
Examples of SBB methods, including links to a more in-depth explanation of how they work, can be found here:
Sequencing by ligation
SBL platforms employ ligase instead of polymerase enzymes to replicate single stranded DNA fragments. Short sequences of nucleotides, tagged with fluorescent markers, are introduced to the sample. The ligase enzyme preferentially joins the sequence that matches the template strand, and its fluorescent signal allows the nucleotide sequence of the template strand to be determined.
Sequencing by oligonucleotide ligation and detection (SOLiD) is an example of SBL, and this video explains in more detail how the SOLiD technique works. Note that SOLiD sequencing is known to struggle with palindromic sequences, as they can form hairpins. The ligase therefore cannot join fluorescently-labeled nucleotide sequences to palindromic regions, and they remain inaccessible.
Long read sequencing
In contrast to short read sequencing approaches, long read sequencing can process DNA fragments that span several thousand base pairs in length. There are various platforms that use distinct methods to extend the read length, and they can be subdivided into ‘true’ and ‘synthetic’ long read technologies.
True long read sequencing
True long read technologies, such as PacBio and Nanopore sequencing, enable the sequencing of long DNA strands in a single continuous process, eliminating the need to fragment them into shorter pieces.
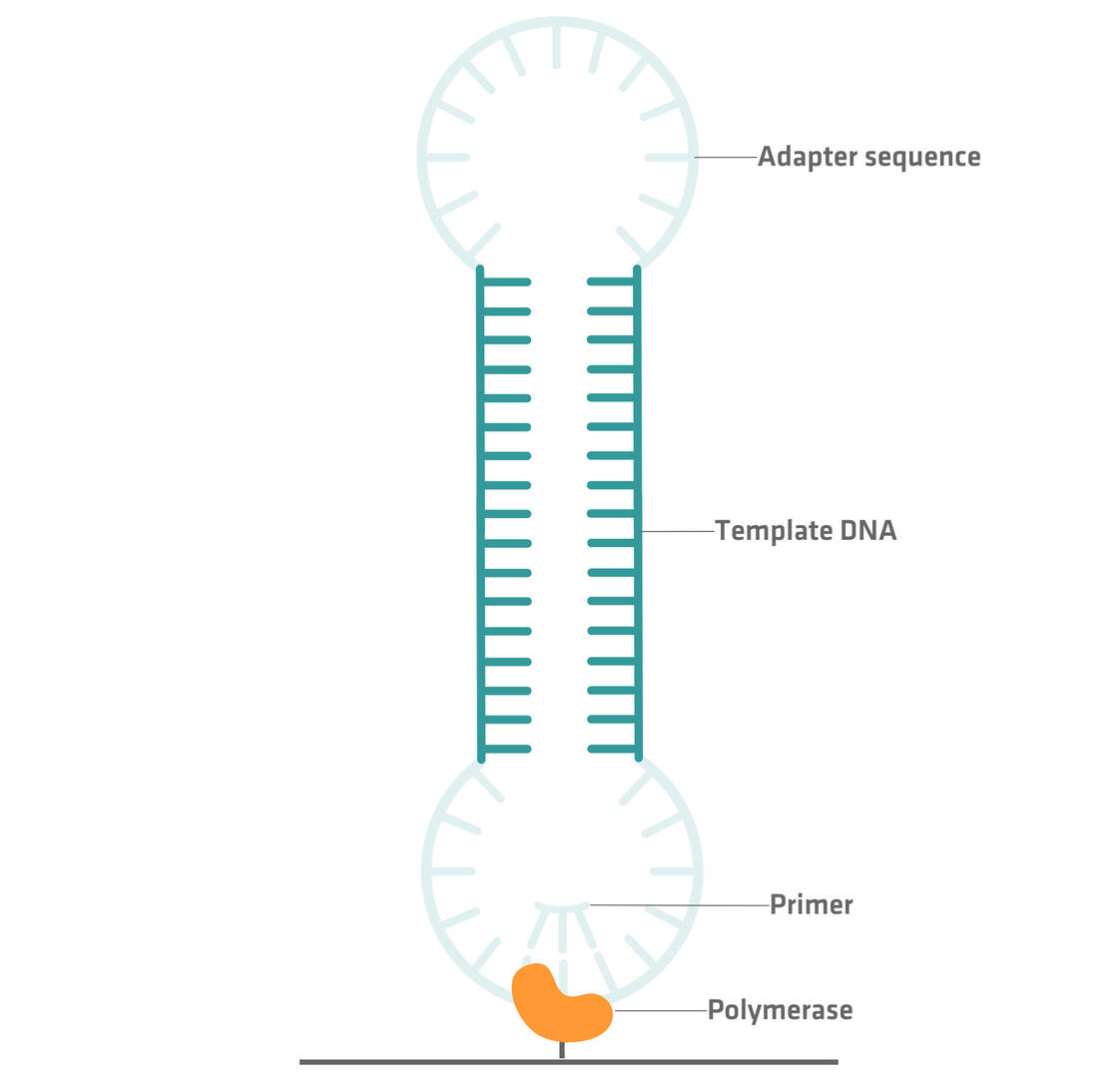
In PacBio sequencing, adapter sequences are ligated to both ends of a double stranded DNA fragment, creating a closed loop. A primer and a polymerase enzyme specific to the adapter sequence are then added to the circular DNA structure, and the polymerase enzyme is immobilized to a solid surface. By monitoring the fluorescent signals of labeled nucleotides incorporated by the enzyme, the nucleotide sequence of the DNA template can be determined.
In Nanopore sequencing, neither polymerase enzymes nor nucleotides are used to sequence DNA. Instead, a constant voltage is applied to a membrane with an array of nanopores. As negatively-charged single stranded DNA molecules travel through the pores, the current across the pores is disrupted by the DNA's nucleotide sequence. These unique variations in the current are precisely interpreted by detectors, enabling the determination of the nucleotide sequence.
More information on Nanopore sequencing
If you wish, you can also watch our video on the true long read sequencing technologies PacBio and Nanopore below.
Synthetic long read sequencing
Several NGS companies that initially developed a short read sequencing technique have subsequently introduced approaches to generate synthetic long reads on their platforms. Unlike true long read sequencing methods that directly sequence DNA fragments several thousand base pairs in length, synthetic long read sequencing techniques computationally reconstruct longer sequences from a collection of shorter reads. This can be achieved by barcoding or landmarking long DNA molecules before or while fragmenting them. These barcodes or landmarks enable the reassembly of the short reads into longer contiguous sequences through specialized sequencing software.
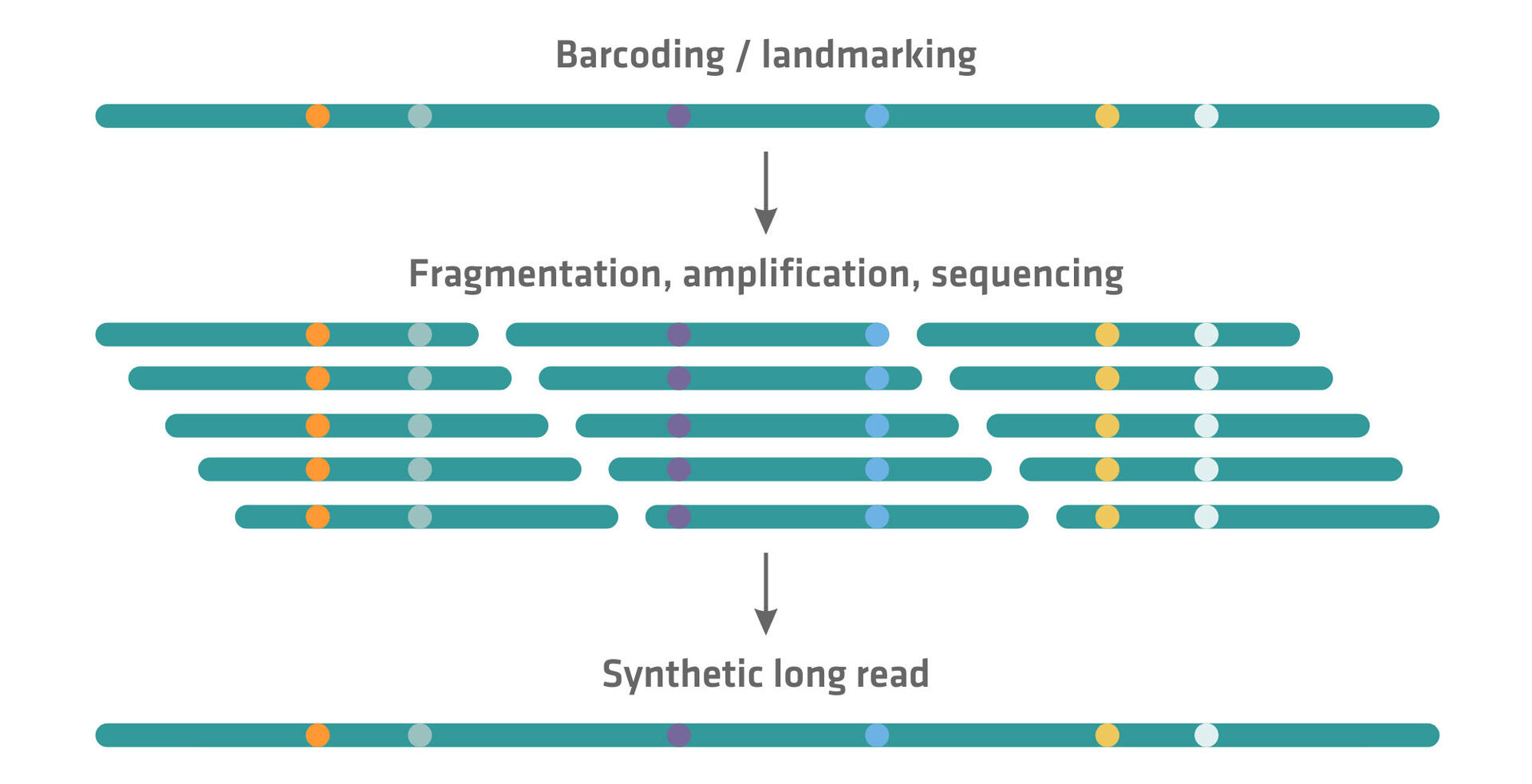
Examples of synthetic long read sequencing methods, including links to a more in-depth explanation of how they work, can be found here:
Advantages and limitations of short read and long read sequencing
Short read and long read sequencing approaches each present unique advantages and limitations that make them suitable for specific applications in the field of genomics.
Short read sequencing techniques are characterized by their ability to accurately sequence DNA fragments ranging from 50 to 300 base pairs, and are the preferred solution for high throughput genomic analyses. As they tend to be more cost-effective per million bases than long read sequencing technologies, they are predominant for applications such as:
- Whole genome sequencing (WGS)
- Whole exome sequencing (WES)
- Gene panel testing
- Single gene testing
These applications benefit from the high accuracy and throughput of short read sequencing, allowing detailed analysis of genetic variations, such as single nucleotide polymorphisms (SNPs), and small insertions or deletions within a genome.
However, the limited read length presents a number of challenges. Complex genomic regions, structural variations and large stretches of repetitive sequences can push short read sequencing methods to their limits, leading to gaps and ambiguities in the assembled sequences. Furthermore, tasks such as de novo assembly, transcriptome analysis (including gene isoform identification), and metagenomics analysis often require longer reads for accurate resolution. True or synthetic long read sequencing is therefore the preferred method for applications requiring comprehensive genomic information, even though it is usually more expensive than short read sequencing in terms of the sequencing cost per base.
The two true long read sequencing methods – PacBio and Nanopore – offer the ability to span entire genomic regions in single reads, and have several further capabilities worth mentioning:
- Nanopore sequencing can directly sequence RNA molecules, without the need for reverse transcription.
- PacBio sequencing can monitor the time lapse between base incorporations, providing insights into base modifications, such as methylation.
We won't further comment on the specific pros and cons of individual short read and long read sequencing techniques here but, if you're interested in a detailed comparison of the prevalent NGS methods, please read this article comparing Illumina, PacBio and Nanopore sequencing for a more comprehensive evaluation.
Conclusion
Ultimately, the decision between short read and long read sequencing is dictated by research goals and genome complexity. Each approach has its place in the genomic toolkit, and offers advantages that can lead to the uncovering of secrets in our DNA.
Did you like this article? Stay tuned for more!
Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.