What is CRISPR-Cas9 and how does it work?
Written by Anina Werner
18. October 2022
The most effective solution to a problem is to solve it at its source rather than simply fighting the consequences. That is exactly how CRISPR-Cas9 could revolutionize medicine. The technique is still in its early stages, but unlike other therapies that treat abnormal proteins associated with a particular disease, CRISPR-Cas9 has the potential to directly alter the genes responsible for the production of these proteins. However, this technique can do much more than just eliminate undesired mutations. Read on to learn more about other applications of CRISPR-Cas9, its mechanism of action, advantages over other genome editing methods, and the challenges that remain.
Table of contents
What is CRISPR-Cas9?
CRISPR-Cas9
CRISPR-Cas9 is a genome editing technology used to change parts of the genome by removing, adding, or altering sections of DNA. It uses a pre-designed RNA sequence to guide the Cas9 enzyme to the location where the genome needs to be altered. The Cas9 enzyme then cleaves the DNA strands, and the cell's natural repair mechanism joins the two ends together by removing, adding, or replacing certain sections.
How does CRISPR-Cas9 work?
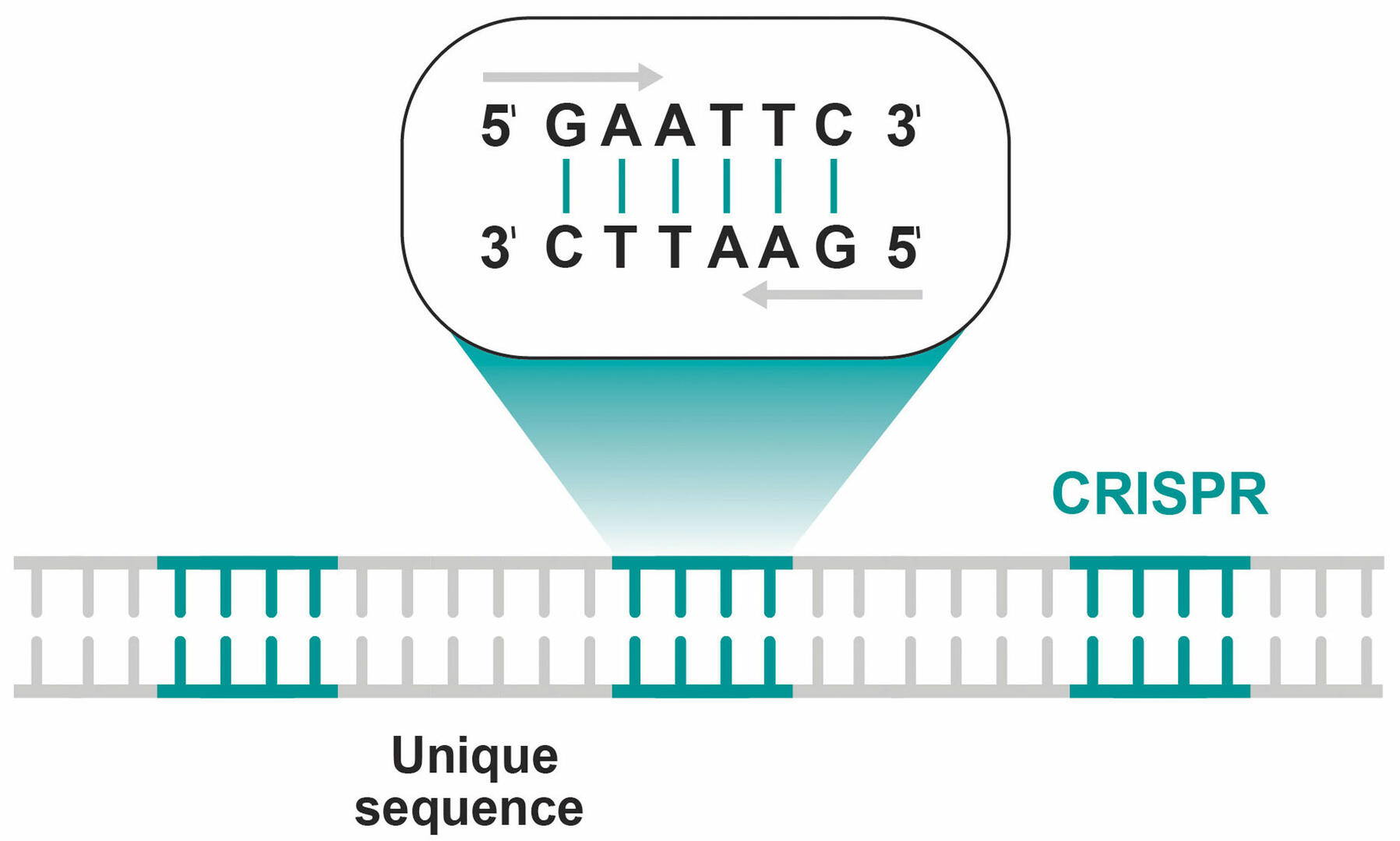
CRISPR-Cas9 has its origin in the immune system of bacteria and archaea. In the 1980s, it was discovered that the genome of E. coli contains DNA sequences known as clustered regularly interspaced short palindromic repeats, abbreviated to CRISPR. These repeating sequences of DNA nucleotides are the same when read from 5' to 3' on the plus and minus strands, and are separated by unique nucleotide combinations (Figure 1).1,2
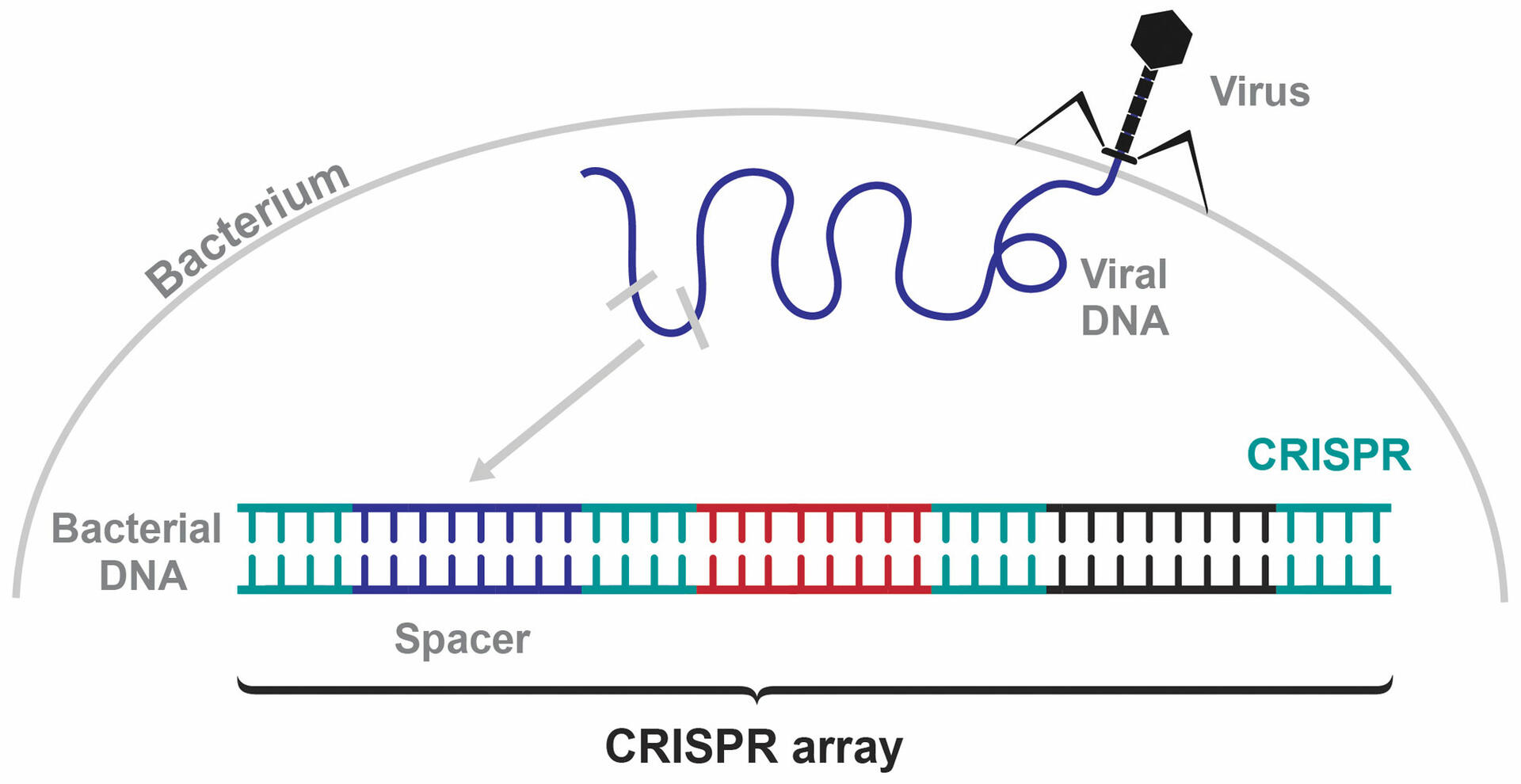
Francis Mojica discovered the bacterial immune system when investigating the DNA sections in between the CRISPR sequences in E. coli. If a bacterium is infected by a bacteriophage, it acquires a small piece of the foreign viral DNA and integrates it into its own genome in between two CRISPR sequences. These variable sequences are called spacers, and the combination of spacers that a bacterium acquires form a CRISPR array (Figure 2).1,2
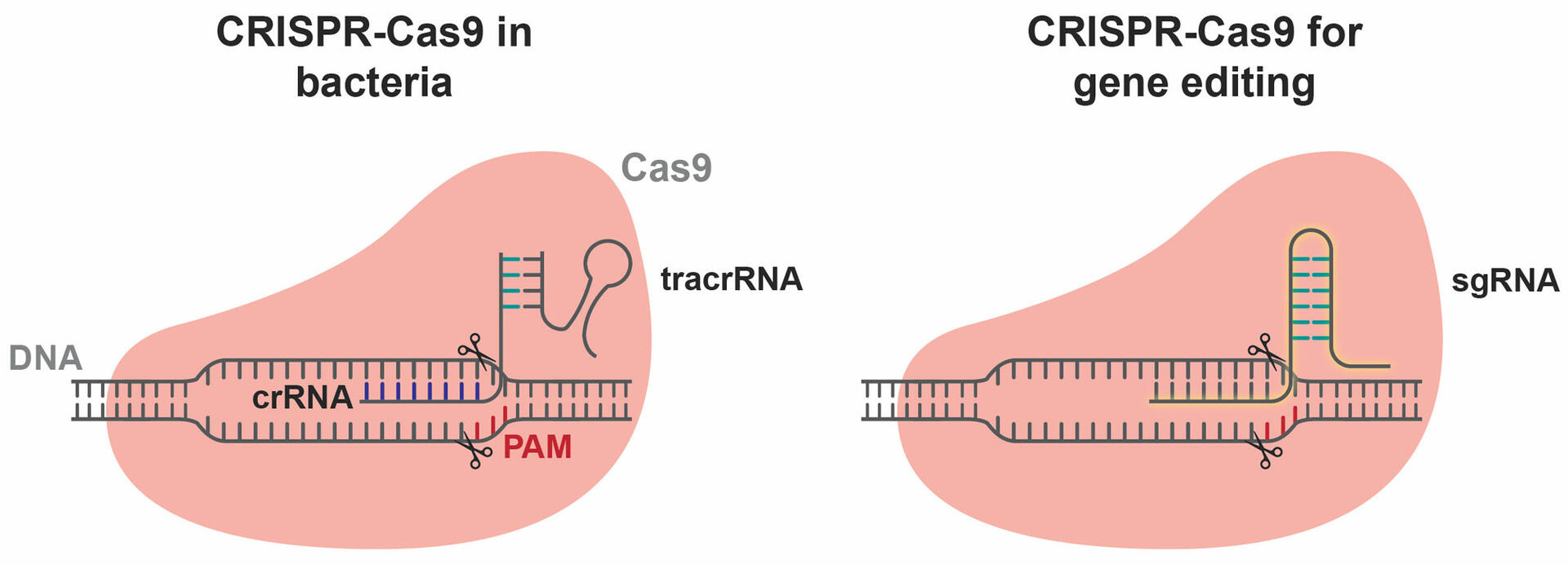
The CRISPR array is part of a CRISPR locus on a bacterial chromosome. This locus includes two additional sequences that are of interest to us. Firstly, one that is transcribed into a trans-activating CRISPR RNA (tracrRNA), and secondly, a sequence that is used to produce a Cas9 enzyme. Together with the transcription of the CRISPR array, these components form complexes consisting of a Cas9 enzyme, a tracrRNA, and a CRISPR RNA (crRNA) sequence (Figure 3).3,4
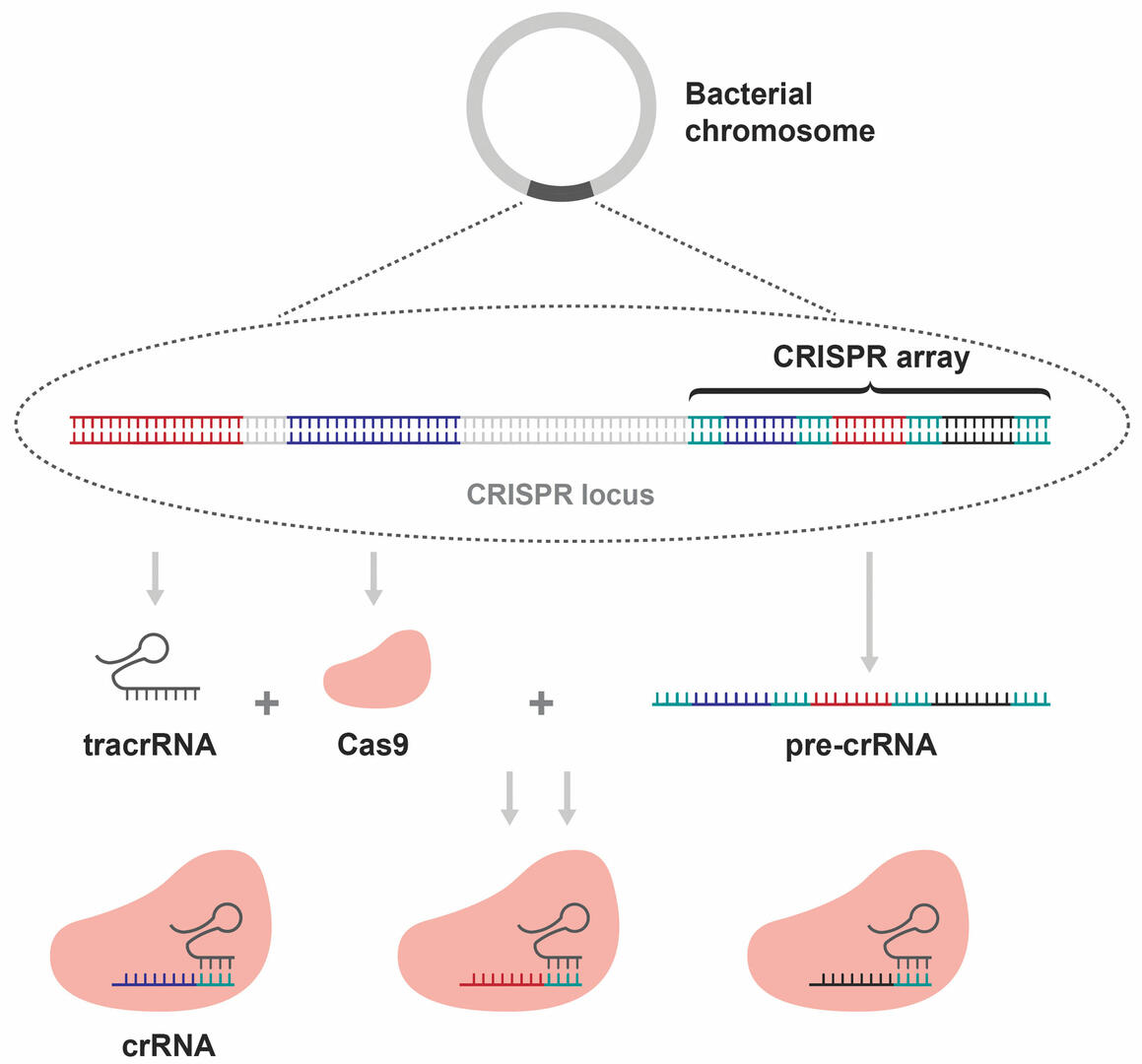
These complexes start looking for the DNA sequences of foreign invaders that are complementary to their crRNA sequence. Once they find a matching sequence, Cas9 cleaves the DNA strands, which consequently protects the bacterium against a second infection by the same bacteriophage.
The detection and cutting process is as follows:
- Cas9 scans the DNA sequences for protospacer adjacent motif (PAM) sequences. PAM sequences are usually 3-5 nucleotides long, and Cas9 enzymes from different bacterial species recognize different PAM sequences based on DNA shape and electrostatics. Let's assume that our bacterium is S. pyogenes, which means that the SpCas9 enzymes will search for a PAM sequence of NGG (where N represents any nucleotide base).
- When the PAM sequence is found, the adjacent DNA segment is unwound and the strand opposite the PAM sequence is compared to the crRNA sequence of the complex. If the sequence is a match, Cas9 cuts both strands of DNA.
- As the viral DNA can no longer be transcribed properly to create new viral particles, the bacteriophage is neutralized.2,5
To ensure that Cas9 only cuts the DNA of foreign invaders and not the CRISPR array that has integrated into the bacterial genome, the repeated CRISPR sequence of a bacterium never contains the PAM sequence that is recognized by the species-specific Cas9 enzyme. For example, as seen above, the SpCas9 enzyme looks for PAM sequences of NGG, whereas the CRISPR sequence in S. pyogenes is coded by GTT, meaning that the enzyme is not able to cut the bacterium's own genome.6
The idea of adapting this bacterial immune mechanism to edit the genome of humans and other organisms was first proposed by Jennifer Doudna and Emmanuelle Charpentier in 2012, who were awarded the Nobel Prize in Chemistry for this research in 2020. To use the CRISPR-Cas9 mechanism as a genome editing technique, the crRNA and tracrRNA molecules are replaced by a single guide RNA (sgRNA) sequence that is synthesized in the lab (Figure 4). When an sgRNA sequence and a Cas9 enzyme are inserted into a cell, the two components automatically form a complex that starts scanning the cell's genome for PAM sequences and sections complementary to the sgRNA, before cleaving the DNA strands.2 The requirement for a PAM sequence is a limitation of CRISPR-Cas9 experiments, but it can often be solved as several Cas9 enzymes are able to recognize multiple PAM sequences. More information on this can be found in the section below, ‘Limitations of CRISPR-Cas9’.
Repair mechanisms
After Cas9 enzymes cut the DNA strands, the cell initiates one of the two repair mechanisms described below.
Non-homologous end joining
If the cell repairs its DNA by the non-homologous end joining (NHEJ) pathway, the two DNA ends are either directly ligated at the cut site or, if the ends are incompatible, processed until a ligatable configuration is achieved. This usually results in nucleotide losses or additions that disable the gene, making NHEJ a great tool for knocking out specific genes, for example, to investigate the impact of a particular gene on an organism’s phenotype. Knocking out a gene can sometimes be a therapeutic option in the clinical field, but in most cases, homology-directed repair (HDR) is better suited for medical purposes.7,8,9
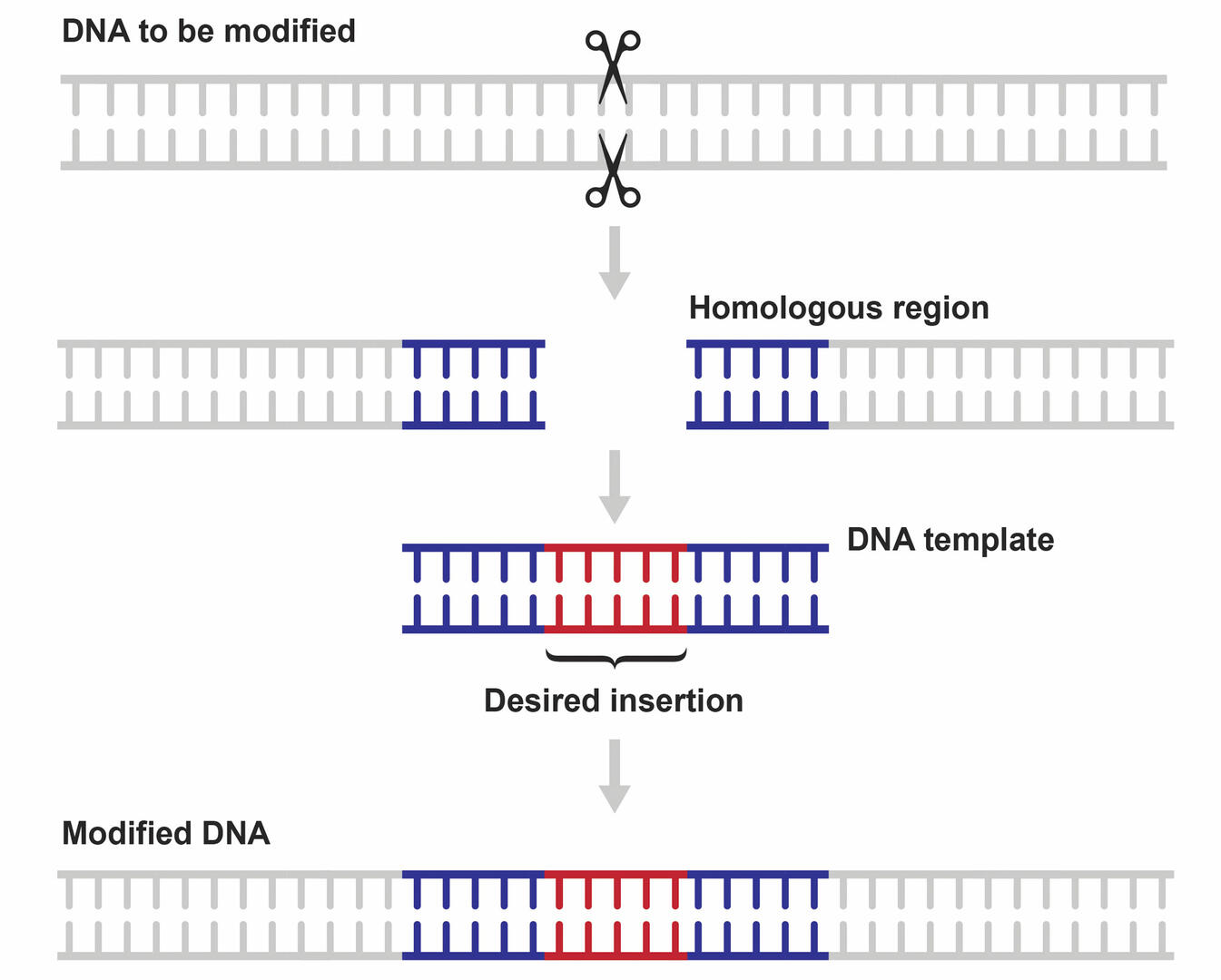
Homology-directed repair
HDR is a repair mechanism that allows scientists to fix double-strand breaks without errors. It requires a DNA template that contains the desired insertion or modification, and segments homologous to the ends of the cleaved strands (Figure 5). As the template will guide the repair process by allowing the cell to ‘copy’ its nucleotide sequence, this repair pathway can be used to add, remove, or change DNA sequences, instead of just knocking genes out.2,9,10 However, as NHEJ is the preferred repair pathway of cells, designing a successful CRISPR-Cas9 application where the double-strand breaks are repaired using HDR can be challenging. Several strategies are available to improve HDR efficiency, and these are described below in the section ‘Limitations of CRISPR-Cas9’.
Delivery
Now that we understand how CRISPR-Cas9 works, we can turn our attention to the delivery of the different components. It can be challenging to successfully introduce them into a cell’s nucleus, and several forms and methods of delivery are available.
Cas9, sgRNA, and the DNA template for HDR can all be delivered as a DNA plasmid containing the sequences for the three components. Alternatively, Cas9 and the sgRNA can enter the cell in the form of mRNA or as a ribonucleoprotein complex, and the DNA template in the form of a ssDNA oligonucleotide.11,12
Even more options are available in terms of delivery methods. If you opt for a viral delivery method, the CRISPR-Cas9 components are packaged in a viral vector. This is the most commonly used technique for in vivo experiments,11 because of the innate ability of viruses to transport their genomes into host cells.13
Additionally, nano delivery methods can transport CRISPR-Cas9 components by replacing the viral vector with lipid or inorganic nanoparticles.11 An advantage of encapsulating the CRISPR-Cas9 components in non-viral vectors is that they lack viral components and consequently reduce the risk of associated severe immune responses.12 However, the internal cellular processes can lead to a degradation of the capsule and prevent the CRISPR-Cas9 components from entering the cell nucleus. A further disadvantage of nano delivery methods is that there are concerns regarding the long-term toxicity of inorganic nanoparticles.11
The third category of delivery options is the use of physical methods. Electroporation, for example, opens pores in the cell membrane via high-voltage electrical currents to allow the CRISPR-Cas9 components to enter the cell. Another physical method is the microinjection of CRISPR-Cas9 components into the cytoplasm or cell nucleus using a needle. Both methods allow the components to easily enter the cell without becoming degraded, but are not suited for all cell types and settings. Mammalian cells, for example, are sensitive to electrical currents and microinjection is not suitable for high throughput applications. Furthermore, neither technique can be used in vivo.11
Advantages of CRISPR-Cas9
Before the invention of CRISPR-Cas9, genome editing techniques focused on zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) (Figure 6). Both techniques require you to design a pair of DNA-binding protein chains attached to a non-specific DNA-cleavage domain of the restriction enzyme FokI. These protein chains either consist of several zinc finger domains that each bind to a specific sequence of three DNA base pairs, or of proteins that bind to a single DNA nucleotide. The two protein chains must be specific for the sequence adjacent to the DNA region where you want to create a double-strand break, with one protein chain binding to the plus strand, and the other one to the minus strand. As soon as both protein chains bind to the target site, the two DNA-cleavage domains dimerize and cut the DNA.14
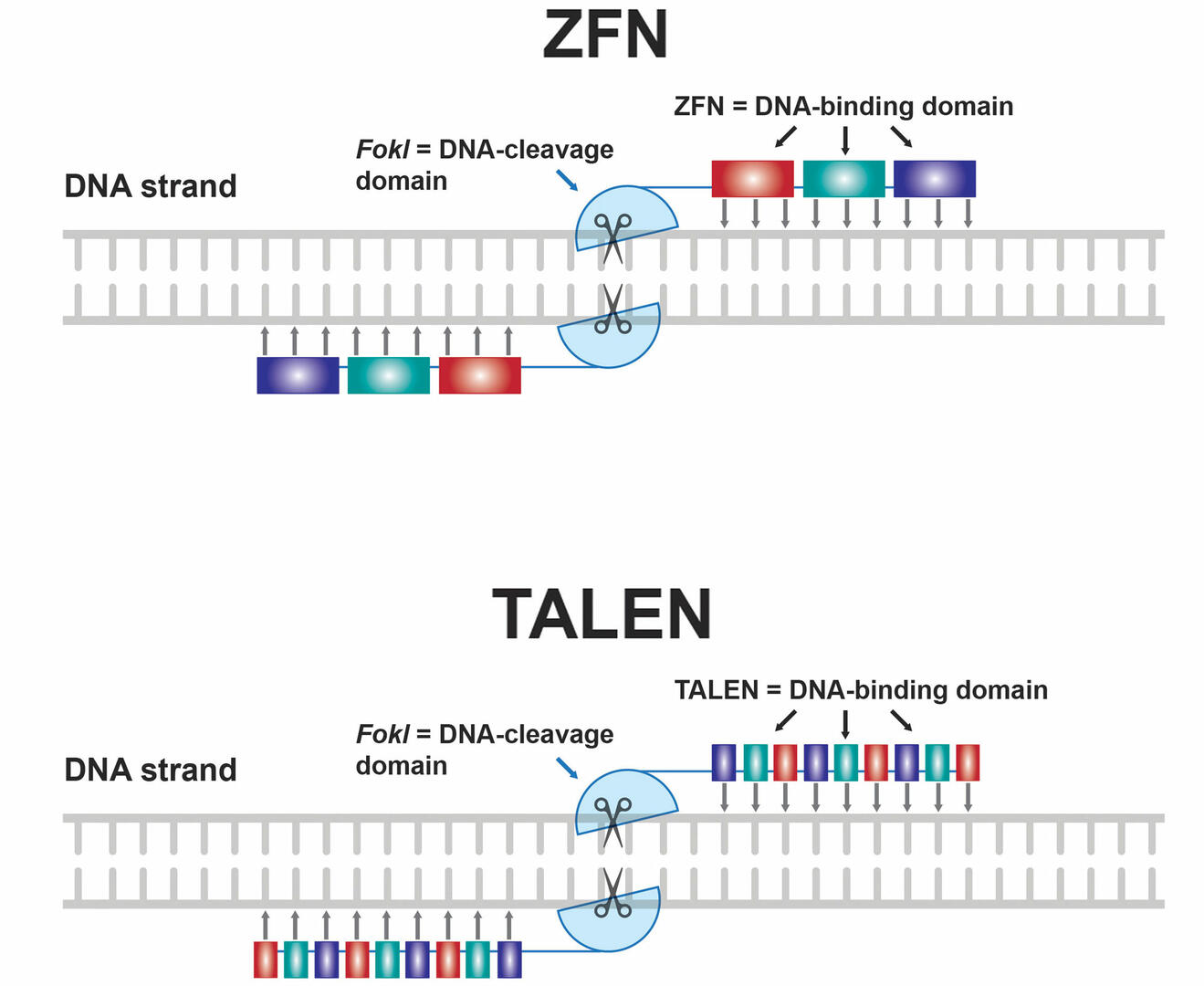
The main advantage of CRISPR-Cas9 over ZFNs and TALENs is that it's less challenging and costly to design an sgRNA that will bind to the genomic site of interest than it is to engineer two protein chains that each recognize a certain target sequence. This means that CRISPR-Cas9 is more flexible, scalable, and user-friendly.15
Limitations of CRISPR-Cas9
Although CRISPR-Cas9 is a very popular and useful gene editing technique, it has several limitations that must also be addressed.
The first limitation is the specificity of CRISPR-Cas9. The sgRNA sequence designed to match the target site is often also partially homologous to genome sequences other than the target sequence. These sequences are called off-target sites; if CRISPR-Cas9 cuts and edits the genome at such a site, this can have unwanted impacts.16 Several strategies are available to reduce off-target effects. You could, for example, redesign your sgRNA to optimize its specificity, or use a high-fidelity Cas9 variant such as SpCas9-HF1, evoCas9, or HiFiCas9. A third option to avoid off-target effects is to induce two single-strand breaks at the same location instead of one double-strand break.9 This can be achieved with Cas9n, an enzyme variant that only cleaves one DNA strand. If you combine two Cas9n enzymes with an sgRNA pair, where one molecule binds to the target sequence on the plus strand and the other one on the minus strand, off-target effects become less likely. This is because double-strand breaks are only produced when both sgRNA molecules bind at the intended location.9,16
The second limitation of CRISPR-Cas9 is its requirement for a PAM near the target sequence. As seen above, Cas9 can only cleave the DNA if a specific 3-5 nucleotide long sequence is located opposite the sgRNA binding site. For the most extensively used Cas9 enzyme, S. pyogenes Cas9, this sequence is NGG. If such a sequence is absent in the genome near the target site, alternative Cas9 enzymes with different PAM sequences must be used.16 A list of common Cas9 enzymes and their PAM sequences can be found here.
Efficiency is another issue to address if you want to use the HDR repair mechanism where the cell fixes the double-strand break by copying a DNA template. As the Cas9 cleavage efficiency is very high when compared to the HDR efficiency, a large number of double-strand breaks will be repaired via the alternative repair mechanism, NHEJ, even if a DNA template is provided.16 Three strategies are available to solve the problem of low HDR efficiency: chemical inhibition, base editing, and prime editing.
Chemical inhibition
HDR efficiency can be improved by chemically inhibiting the NHEJ pathway. However, as the agents needed to suppress the NHEJ repair mechanism are harmful, this solution is sometimes unsuitable, for example, in clinical applications.9
Base editing
The HDR efficiency issue can also be solved by not cutting the DNA strands at all. Catalytically inactive dCas9 – an enzyme that can still bind to a DNA sequence but no longer cut it – fused to a deaminase can be used to create cytosine and adenine base editors.9 Instead of cutting the DNA strands, these base editors convert a C-G base pair into a T-A base pair, or an A-T base pair into a G-C base pair, making them a useful alternative for single-base gene editing.17
Prime editing
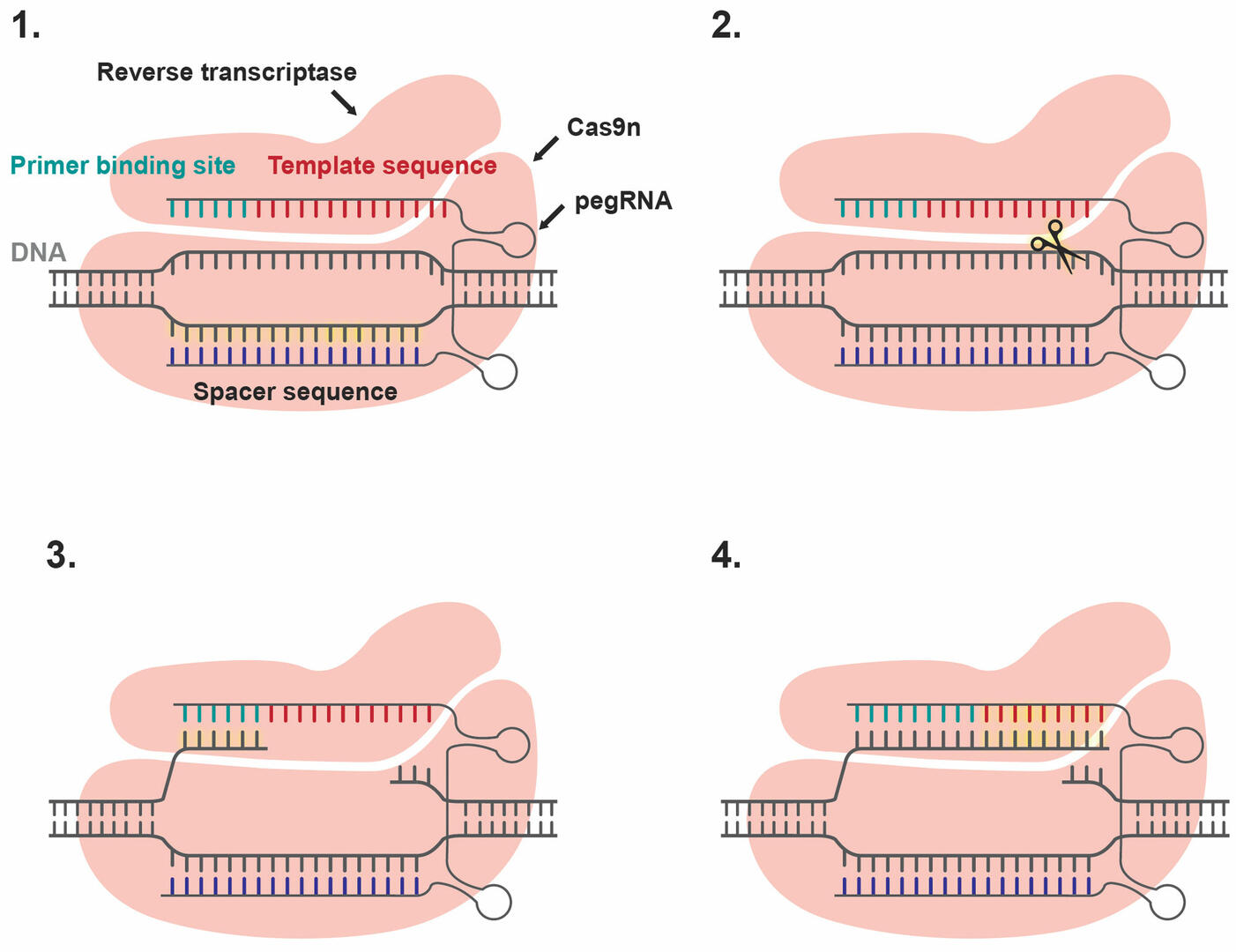
If base editing is not an option because a larger DNA sequence needs to be added, removed, or altered, prime editing might be a useful alternative. It uses a Cas9n enzyme fused to a reverse transcriptase and a prime editing guide RNA (pegRNA). The pegRNA contains a spacer sequence, a primer binding site, and a template sequence with the gene edit that you want to make. The spacer sequence is responsible for guiding the Cas9n enzyme to the site where the genome needs to be edited (Figure 7, Step 1). Once it has arrived, Cas9n cuts the strand opposite the spacer sequence (Figure 7, Step 2). The primer binding site of the pegRNA then anneals to one of the snipped ends (Figure 7, Step 3), and reverse transcriptase inserts the desired edit by copying the template sequence (Figure 7, Step 4). The cell has two options to repair the single-strand break. It can either seal in the edited sequence and trim off the old fragment, or vice versa. Unfortunately, techniques to improve the odds of the cell choosing to seal in the edited sequence still need to be developed.18 If the cell fixes the single-strand break by sealing in the edited strand, there will be a mismatch between the plus and minus strand. To increase the chances of the cell repairing this mismatch by editing the unaltered strand to match the edited strand and not the other way around, you can produce a single-strand break in the unaltered strand using an sgRNA-Cas9n complex.18,19
Applications
The broadest range of applications for CRISPR-Cas9 can be found in the clinical field, as the technique has the potential to cure genetic diseases such as blood disorders, cancer, neurodegenerative diseases, and much more.20 Vertex Pharmaceuticals and CRISPR Therapeutics, for example, could submit a gene editing medicine based on CRISPR-Cas9 for approval in the US, the UK and Europe before the end of the year. Clinical trials of the therapy have been conducted with patients with sickle cell disease – a genetic mutation resulting in crescent-shaped red blood cells that can cause vaso-occlusive crises by blocking the blood flow – and patients with beta thalassemia, a blood disorder reducing the production of hemoglobin. Trial data suggests that the therapy can completely eliminate vaso-occlusive crises caused by sickle cell disease, and allow patients with beta thalassemia to stop blood transfusions, or at least to considerably reduce the number of required transfusions. If the therapy gains approval, it would become the first marketed therapy based on CRISPR-Cas9.21 CRISPR-Cas9 could even be used for germline editing of human embryos. However, as ethical concerns and the risks of creating gene edits that are passed on to future generations outweigh the benefits, it's illegal to use CRISPR-Cas9 for germline editing in most countries.22,23
CRISPR-Cas9 also has great potential in agriculture, where it could be employed to make crops more resistant to certain diseases or increase their environmental stress tolerance to help feed the growing global population. It could also allow us to produce healthier food.24,25,26 Examples of 'CRISPR-Cas9 foods' include tomatoes containing a high level of an amino acid that is believed to aid relaxation and lowering blood pressure – which are currently sold in Japan25 – and a strain of wheat that has been grown in field trials in the UK with the aim of reducing levels of the carcinogen asparagine in bread.26
Moreover, CRISPR-Cas9 could help us to tackle climate change, for example, by improving the tolerance of yeast to the production conditions of biofuels,20 or by manipulating the yeast to transform sugar into hydrocarbons that can be used to make plastic without relying on petroleum.24
Conclusion
As you can see, CRISPR-Cas9 is a useful technique for a vast array of applications, and it will be interesting to see what this relatively new gene editing method will be capable of achieving in the coming years and decades.
What do you think will be possible in the future, thanks to CRISPR-Cas9? Let us know your thoughts in the comments section below.
Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.