
ELISA vs western blot: a comparison of two common immunoassays
Written by Éva Mészáros
02. November 2022
If you work in a laboratory and perform immunoassays, you have probably had to choose between the enzyme-linked immunosorbent assay (ELISA) and western blot techniques at some point. Both approaches have their pros and cons, so deciding which one to use can be challenging. This blog post takes a closer look at these techniques, to help you select the most appropriate method for your lab’s workflow.
Table of contents
What are immunoassays?
Immunoassays rely on the antibody-antigen binding mechanism that occurs naturally in the immune system. An antibody produced by the adaptive immune response is specific to a certain antigen only, and immunoassays take advantage of this specificity to identify a molecule of interest – typically a protein – in a sample.
Many different types of immunoassays have been developed over the last few decades, including the ELISA and western blot, two common methods that we will compare and contrast in this article.
ELISA
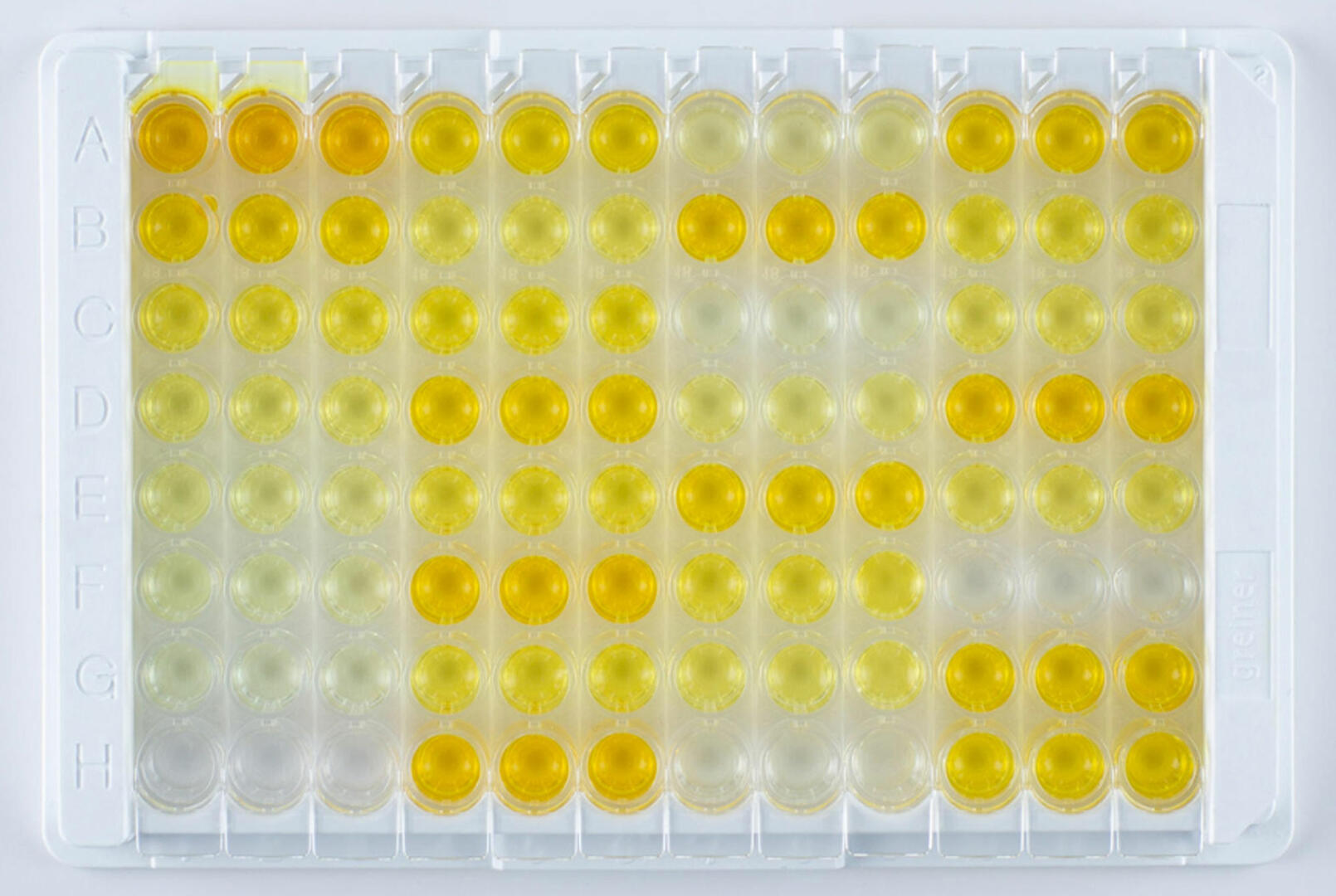
ELISAs are highly specific and sensitive assays used to detect concentrations of as little as 0.01 nanograms of antigen or antibody per milliliter of sample.1 The four main types of ELISA – direct, indirect, sandwich, and competitive – are all usually performed in 96 well plates, using the bottom of the wells as a solid surface to immobilize antigen-antibody complexes. Since an enzyme is covalently attached to one of the molecules in the complex, the subsequent addition of an enzyme-specific substrate results in a detectable colored reaction product if the antigen or antibody of interest is present in the sample. ELISAs can either deliver qualitative, semi-quantitative or quantitative results.
To learn more about how the four different types of ELISA work, their advantages and disadvantages, as well as data analysis, please refer to our blog post 'An introduction to the different types of ELISA tests'.
Western blot
Before delving deeper into the details and methodologies involved in western blotting, we’d like to share the story behind its name with you.
The western blot was developed in 1981 by Walter Neal Burnette, a postdoc in the Nowinski lab at the Fred Hutchinson Cancer Center in Seattle. The name 'western blot' alluded to the Southern blot invented in 1975 by Ed Southern, and the northern blot invented in 1977 by James Alwine. He chose the ‘western’ and not ‘eastern’ direction descriptor, to make a geographical reference to the location of his lab in Seattle, on the West Coast of the United States.2,3
Now that you know the origin of its name, let's have a look at how this technique works.
What is a western blot?
In a nutshell, the western blot (also called an immunoblot) is a technique used to detect a specific protein, such as an antigen, in a sample consisting of a mixture of proteins. To this end, the proteins in the sample first need to be denatured and separated by size.
After transferring the proteins to a membrane, antibodies are used to detect the protein of interest, and the result is visualized using a colorimetric, chemiluminescence or fluorescence analysis method. Radioactive detection is also possible, but is now only rarely used due to health and safety risks.
For those who aren't familiar with the technique, we've explained the various parts of western blotting in more detail below.
Steps of a western blot
We can break western blots down into three main steps:
- Separation of proteins by size
- Transfer of proteins to a membrane
- Labeling of proteins using antibodies
The first step is achieved using gel electrophoresis. Samples are loaded into wells on top of the gel. An electrical current is then applied to the gel, causing the proteins to travel through it. Two factors influence how fast the proteins in the sample travel: size and charge. To separate the proteins according to their size, they need to have a proportionally uniform charge, therefore, they are treated with sodium dodecyl sulfate (SDS) which causes proteins to unfold into linear chains and imparts a negative charge.4
The porous polyacrylamide composition of the gel allows smaller proteins to migrate faster than larger ones, resulting in the formation of bands containing proteins of the same size. The polyacrylamide concentration determines the size of pores in the web of the gel, and can be adjusted to ensure even distribution of proteins and a satisfactory electrophoresis result. Generally, larger proteins separate easily in a gel with a low percentage of polyacrylamide, and smaller proteins require a gel with a high percentage of polyacrylamide. Please note that unpolymerized acrylamide is carcinogenic and mutagenic, and should always be handled in a biosafety cabinet using appropriate personal protective equipment.
In addition to your samples, you also need to add a molecular marker containing several proteins with known molecular weights to the gel. This will allow you to see if electrophoresis has been successful, and whether the subsequent transfer of the proteins to the blotting membrane has been effective. Moreover, it will enable you to estimate the length of your proteins of interest, as you can compare their migration path through the gel with the distance travelled by the proteins of the molecular marker. The method is often abbreviated as SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) because gel electrophoresis for western blots uses SDS and polyacrylamide.

The second stage of the western blotting process involves transferring the migrated proteins to a blotting membrane. The most commonly used technique is electrophoretic transfer, where the gel and membrane are sandwiched between filter paper and placed between electrodes, after which an electric field moves the proteins from the gel to the membrane.5
The third step of a western blot protocol – protein labeling – is very similar to an ELISA. It requires you to incubate the membrane with a blocking buffer, which binds to any remaining protein binding sites to reduce subsequent non-specific binding of antibodies to the membrane. After washing the membrane you can opt for either a direct or indirect labeling method.
In the direct method, the membrane is incubated with labeled antibodies that bind to the proteins of interest, before washing unbound antibodies away. The indirect method uses two antibodies, a primary and a secondary antibody. In the first step, the membrane is incubated with unlabeled primary antibodies that bind to the proteins of interest. After washing, a second incubation is performed, using labeled secondary antibodies that bind to the primary antibodies. This is followed by a final washing step to remove any unbound molecules.
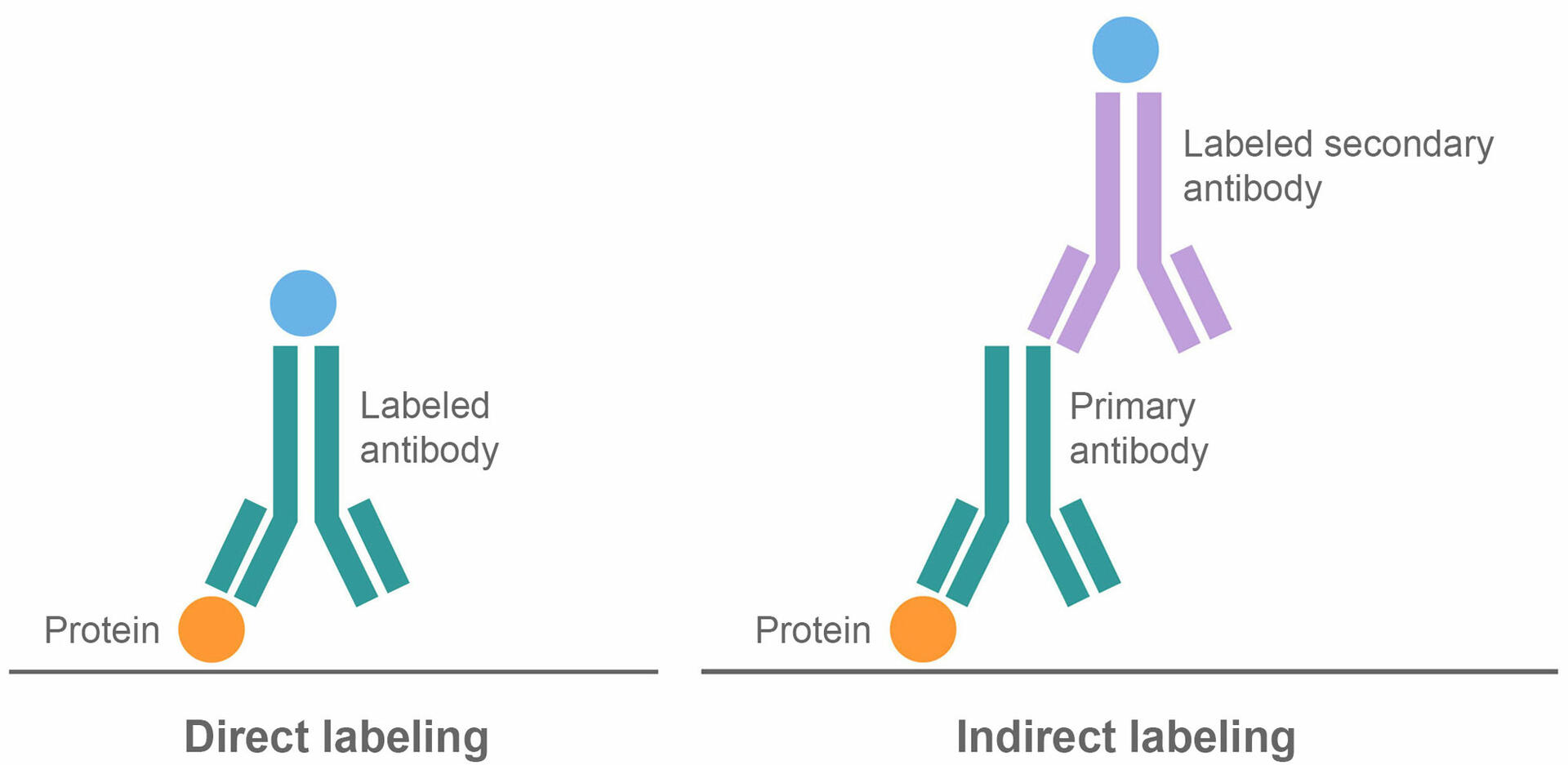
There are risks and benefits to both labeling protocols. Direct binding is a shorter process, which can save time and reagents. It also reduces the risk of non-specific antibody binding since it only uses one antibody.
On the other hand, indirect western blots are more sensitive because several secondary antibodies can bind to one primary antibody, amplifying the detectable signal. The indirect method also offers greater flexibility, as secondary antibodies can be specific to several primary antibodies of the same type and from the same host species. This means that the same labeled secondary antibody can be used in different indirect western blot applications, whereas several labeled primary antibodies are needed to perform different direct protocols.
Western blot analysis
To analyze a western blot, you need to measure the signal produced by the labeled antibodies. The detection steps and equipment differ depending on whether the antibodies are tagged with an enzyme or a fluorophore.
Enzymatic detection
You can use either colorimetric or chemiluminescence detection methods to analyze your western blot if your antibody is labeled with an enzyme.
Colorimetric methods
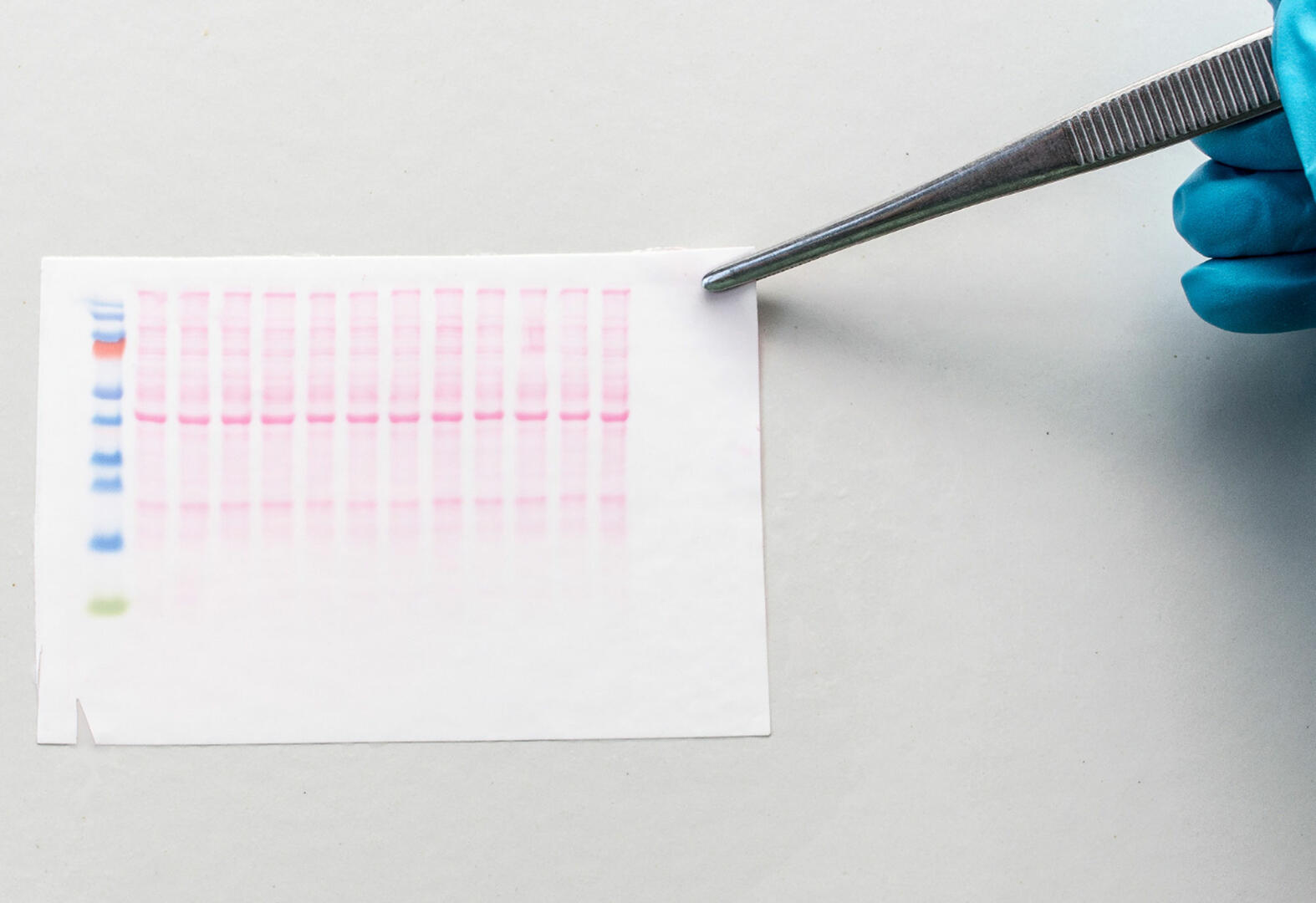
Colorimetric methods require the application of an enzyme-specific chromogenic substrate to the membrane; the reaction catalyzed by the enzymes attached to the antibodies will produce a colored reaction product. A stop solution is added to terminate the color development, and the chromogenic products of the enzymatic reaction are visible as bands to the naked eye.6
A colorimetric western blot analysis can be performed quickly and cost-effectively because you don't need any special detection equipment. It is, however, not very sensitive. Proteins in the nanogram range are required to produce a visible band.6
Chemiluminescence methods
Chemiluminescence detection methods also require an enzyme-specific substrate, but one that is luminescent instead of colorimetric. This means that the enzymatic reaction on adding substrate to the membrane will produce light as a by-product. The light can then be detected using an X-ray film or a charge-coupled device camera (CCD).6
The main advantage of chemiluminescence detection is its sensitivity; it can be used to detect and quantify as little as femtograms of the protein of interest. However, a downside of this technique is that light is only emitted briefly, when the enzyme converts the substrate into a product, and therefore detection needs to be performed immediately after substrate addition.7
Fluorescence detection
If your antibodies are labeled with fluorophores, you don't need to add any substrate to the membrane. Instead, you can image the membrane directly using a fluorescence imaging system, where a light source excites the fluorophores and the fluorescent signals emitted are detected by wavelength-specific filters and digital camera systems.8
Tagging antibodies with fluorophores instead of enzymes has several advantages. Firstly, tagging individual antibodies with different fluorophores allows for the simultaneous detection of various proteins with similar molecular weights (multiplexing). Secondly, as fluorescent signals are still detectable on membranes after months of storage at room temperature, this method offers a lot of flexibility when it comes to analyzing assay data. Thirdly, fluorescence detection is simple to perform because you don't need to add any substrate. The only disadvantages of this detection method are the need for a special imaging system, and reduced sensitivity compared to chemiluminescence detection.7
ELISA vs western blot
Now that you understand how to conduct and analyze the two immunoassays, how should you decide which technique to use?
Generally speaking, an ELISA is easier and faster to perform than a western blot because the protocol is shorter. Moreover, ELISAs are better suited for high throughput labs because they can be performed with lower sample volumes and are usually carried out in 96 well plates, allowing for workflow automation.

When it comes to obtaining reliable quantitative data in an ELISA, a simple standard curve – serial dilutions of a known target protein – can be set up, and its equation can be used to calculate the antigen or antibody concentration of samples based on their absorbance values. More information on how to set up quantitative ELISAs can be found here.
Performing a quantitative western blot can be more demanding. It requires the consideration of several factors, such as overloading of samples, membrane saturation and signal saturation. Additionally, internal loading controls like a housekeeping protein are required, data must be normalized, and the background signal must be subtracted for quantitative analysis. More information on how to set up quantitative western blots can be found here.
However, even though western blots can be complex and time-intensive, they are preferred over ELISAs for some applications. The first advantage of a western blot is that they are highly specific, and are sometimes used to rule out false positives and confirm positive ELISA results in clinical settings. This is due to the initial protein size separation which ensures that non-specific signals from analytes – which would likely be a different size – can be easily detected as a second band on the membrane.
Secondly, western blots generate more information about the target protein and the sample in general. Rather than simply identifying the presence or absence of an analyte in a sample, western blots can also determine the length of a protein and analyze sample purity. Therefore, the slightly longer and more tedious protocol may be worthwhile if your assay requires more extensive information.
A third advantage of western blots – although only true if the detection antibody is labeled with a fluorophore – is its ability to detect several proteins per sample in parallel.
Alternatives to ELISAs and western blots
We have determined that the ELISA and western blot methodologies have their individual benefits and disadvantages for various laboratory applications. However, these techniques fall short when looking to perform more complex high throughput multiplexing immunoassays: the enzyme-labeled antibodies used in ELISAs don't allow you to detect several analytes in parallel, and fluorescent western blots are only suitable for low throughput settings. Luckily, alternative techniques like bead-based immunoassays and protein microarrays have been developed for just this purpose.
Bead-based immunoassays
To perform a bead-based immunoassay, you need as many bead sets as you have analytes of interest. Each bead set is coated with capture antibodies specific for a certain analyte, and labeled with a red fluorescent dye with a unique intensity. Different detection antibodies are also needed for this assay. These are labeled with a green fluorophore, and bind to the analytes of interest. When the bead sets and detection antibodies are added to a sample and incubated, they form complexes with the analytes of interest.
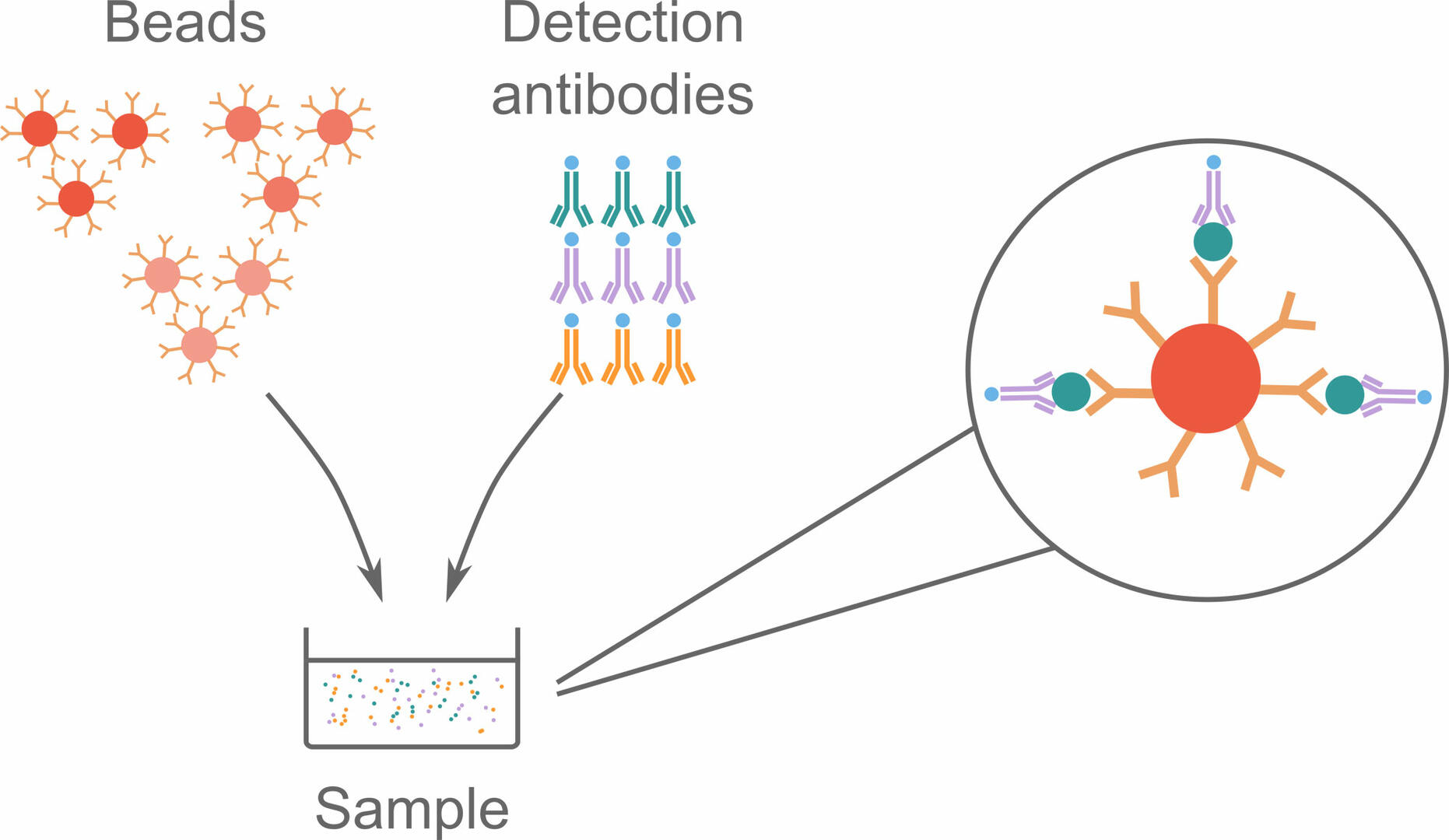
These bead complexes can then be analyzed using flow cytometry, during which individual complexes pass through a red and a green laser beam. The red laser identifies the signal intensity of the fluorescent dye, and can consequently tell which type of analyte the bead is supposed to bind. The green laser is then used to determine whether analytes and detection antibodies are bound to the beads, by detecting fluorophores on the detection antibodies.9,10,11
Protein microarrays
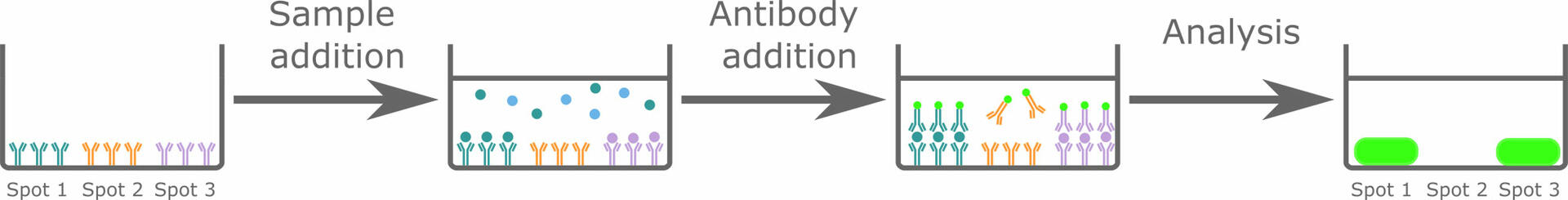
Protein microarrays (also called protein chips) are comparable to performing a lot of sandwich ELISAs simultaneously on a single solid surface. The solid surface – which can be a membrane, a glass slide or the well of a microplate – is pre-spotted with different capture antibodies specific for the analytes of interest. When incubating the solid surface with a sample, the analytes, if present, will bind to the capture antibodies and can then be detected with labeled antibodies. As you know that spot 1 is supposed to bind analyte A, spot 2 analyte B, etc., the pattern generated after analysis will allow you to determine which analytes are present in your sample.
Conclusion
In conclusion, ELISAs and western blots are two immunoassays that can be used to detect the presence of a protein in a sample. Each method has its pros and cons; ELISAs are easier, faster to perform, and better suited for high throughput labs, while western blots are more specific, can provide additional information about the target protein and the sample under analysis, and allow for multiplexing. The decision as to which method to use depends on the application. Moreover, there are alternative techniques such as bead-based immunoassays and protein microarrays that may be more suitable when performing high throughput multiplexing assays.
Did you like this article? Subscribe to our blog for more!
Further reading:



