How to design primers for PCR
Written by Éva Mészáros
24. March 2022
PCR is one of the most widespread molecular biology applications, yet it is anything but simple to perform. Common issues – such as a low product yield or non-specific amplification – are often caused by poorly designed PCR primers. We have therefore summarized the most important information on designing PCR primers to help you overcome these challenges.
Table of contents
What are primers?
Primers
Primers – also called oligonucleotides or oligos – are short, single-stranded nucleic acids used in the initiation of DNA synthesis. During PCR reactions, they anneal to the plus and minus strands of the template DNA, flanking the sequence that needs to be amplified.
If you want to learn more about PCR and primers in general, refer to our blog post: The complete guide to PCR. It explains in more detail what PCR is, what a master mix consists of, and how the different components interact during the three steps of PCR.
How to design PCR primers?
PCR primers have to be tailored to both the region of interest of your template DNA and your reaction conditions. This means that, unlike the other components of the PCR master mix, you can't just buy them, but need to design them yourself. Usually a primer design tool is used. Examples of free primer design tools are:
- Primer-BLAST from the US National Center for Biotechnology Information
- Primer3Plus from the Bioinformatics Group of Wageningen University
- PrimerQuestTM from Integrated DNA Technologies
These tools allow you to set parameters such as primer length, melting temperature, GC content and more. Watch the video below or read on to learn what the optimal values for each of these parameters are, and how they affect your PCR assay.
Primer length
The optimal length of a PCR primer lies between 18 and 24 bp. Longer primers are less efficient during the annealing step, resulting in a lower amount of PCR product. Conversely, shorter primers are less specific during the annealing phase, leading to more non-specific binding and amplification. However, there are exceptions to this rule. For example, some scientists have successfully used miniprimers that are 10 bp long to expand the scope of detectable sequences in microbial ecology assays.
Target sequence length
The target sequence to be amplified should ideally be between 100 and 3000 bp for standard PCR assays, and 75 and 150 bp for qPCR assays. Longer sequences usually need special enzymes and reaction conditions to ensure that they are completely and specifically amplified.
Primer melting temperature
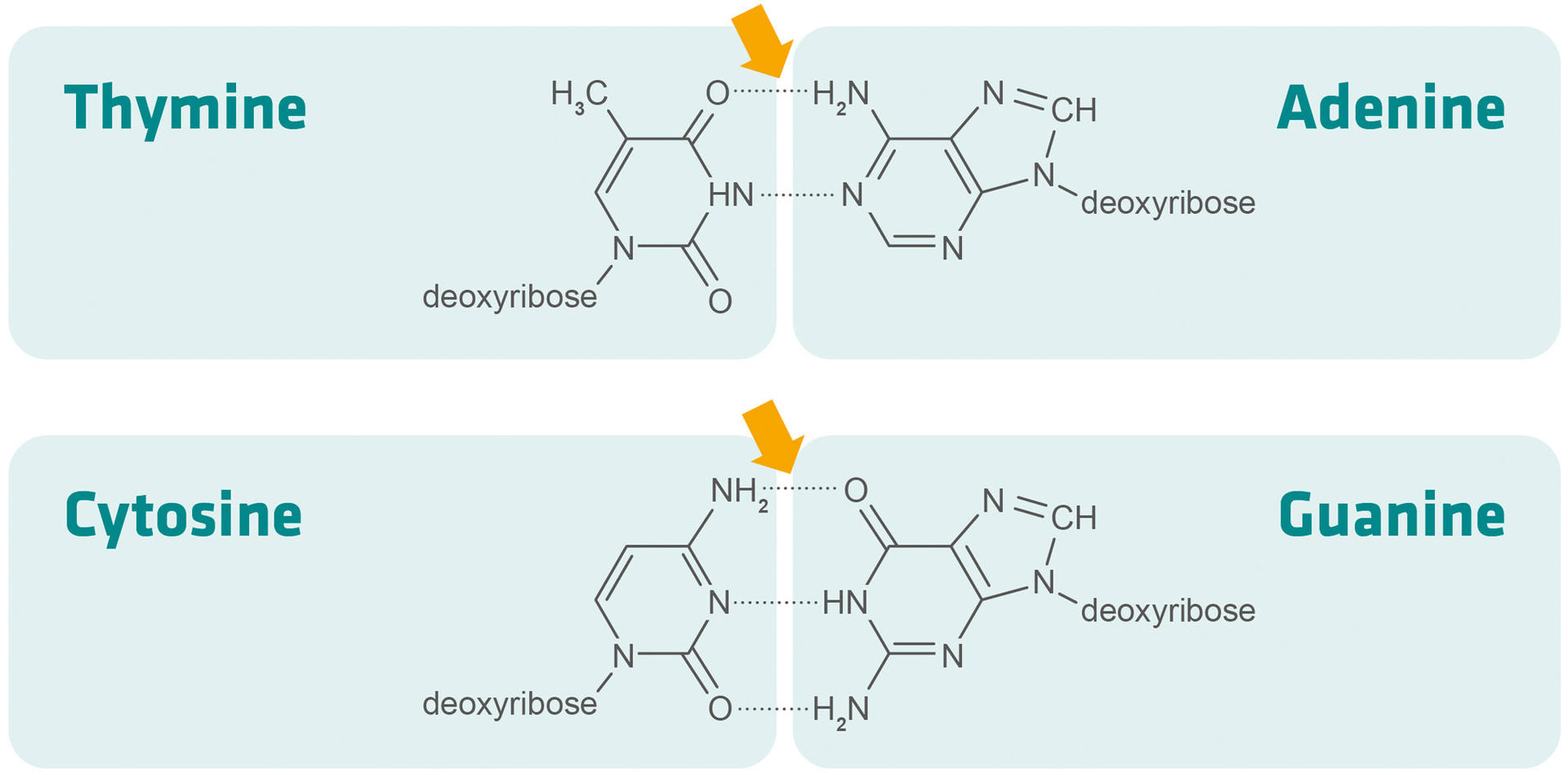
The primer melting temperature (Tm) can be defined as the temperature at which half of the primers dissociate from the template DNA. It is usually between 50 and 60 °C, and the melting temperatures of the forward and reverse primers should be within 5 °C of each other. If the two melting temperatures are further apart, it won't be possible to find an annealing temperature that allows both primers to bind to the template DNA.
Most primer design tools use the nearest neighbor method to calculate primer melting temperatures, as it's the most accurate. However, if you want to make an approximate calculation yourself, you can use this formula:
Tm = 4 °C x (G+C) + 2 °C x (A+T)
Tm: melting temperature
G, C, A, T: number of nucleobases (guanine, cytosine, adenine, thymine) in the primer
As indicated in the formula above, G-C bonds are harder to break than A-T bonds – because G-C base pairs are linked by three hydrogen bonds, and A-T base pairs by two – and the length of the primer also impacts its melting temperature. This means that you can either increase the GC content of a primer (provided the template allows for this), or slightly extend its length if its melting temperature is too low.
Primer annealing temperature
The primer annealing temperature (Ta) is the temperature needed for the annealing step of the PCR reaction to allow the primers to bind to the template DNA. The theoretical annealing temperature can be calculated as follows:
Ta = 0.3 x Tm(primer) + 0.7 x Tm(product) – 14.9
Ta: primer annealing temperature
Tm(primer): lower melting temperature of the primer pair
Tm(product): melting temperature of the PCR product
Once you've calculated the theoretical annealing temperature, the optimal annealing temperature needs to be determined empirically. To achieve this, perform a gradient PCR, starting a few degrees below the calculated annealing temperature, and ending a few degrees above. After amplification, run a gel, and the sample producing the clearest band contains the largest quantity of PCR product, making its annealing temperature the optimal one for your primers. Usually, you'll get a value that is 5 to 10 °C lower than the primer melting temperature.
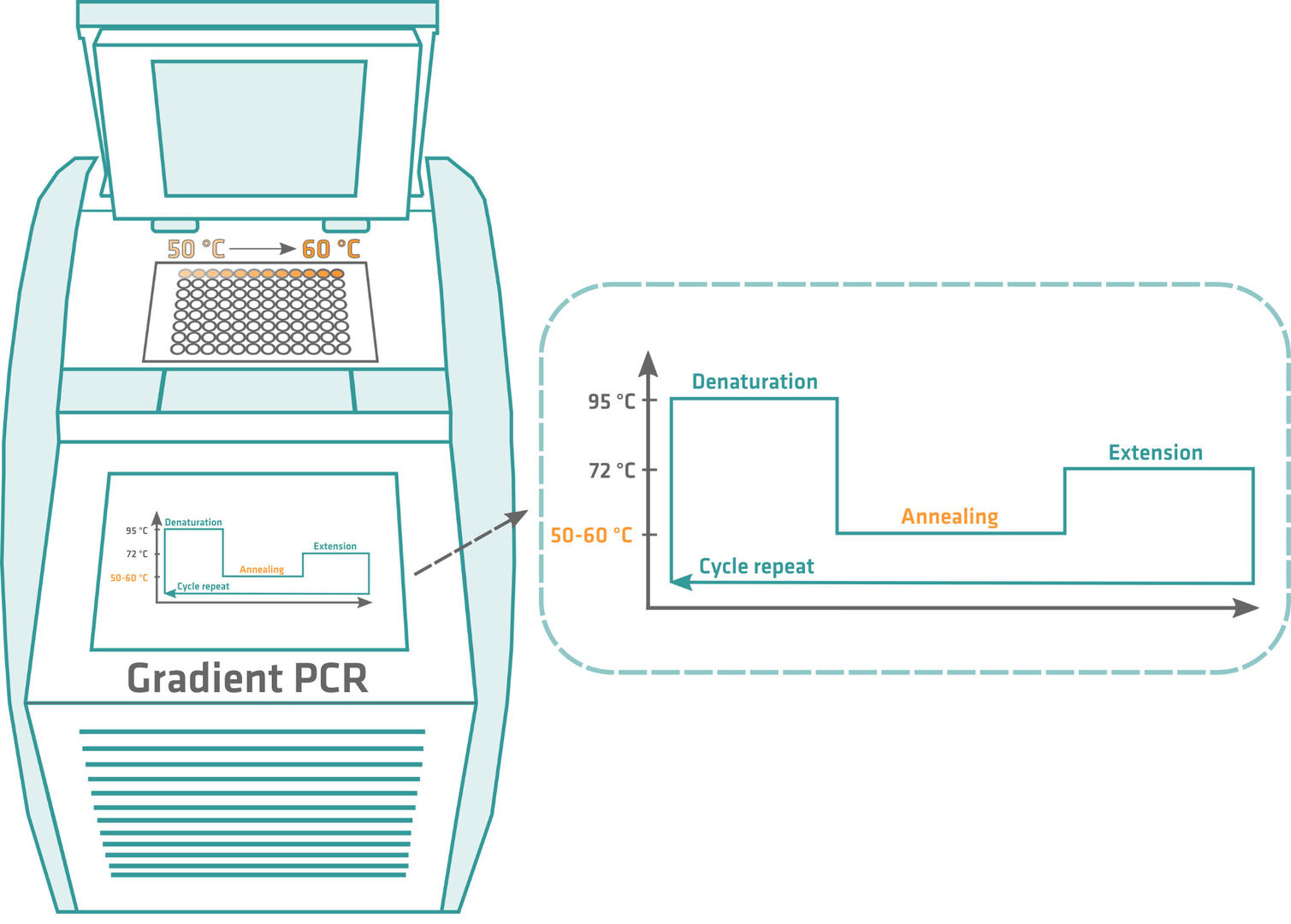
It's important to determine the optimal annealing temperature, as primers could form hairpins or bind to regions outside the DNA sequence of interest if it's too low, producing non-specific and inaccurate PCR products. If the annealing temperature is too high, the primers won't sufficiently bind to the template DNA, and you'll obtain little to zero amplicons.
GC content
As seen before, G-C base pairs are stronger than A-T base pairs, which means that a higher GC content ensures a more stable binding between the primers and the template DNA. The optimal GC content of a primer lies between 40 and 60 %, and primers should have two to three Gs and Cs at the 3' end to bind more specifically to the template DNA.
Runs and repeats
Avoid runs of four or more single bases – such as ACCCCC – or dinucleotide repeats (for example, ATATATATAT), as they can cause mispriming.
Cross homology
If a primer is homologous to a template DNA sequence outside the region of interest, these sequences will be amplified too. Therefore, you should always test the specificity of your primer design against genetic databases; for example, by ‘blasting’ them through NCBI BLAST software.
Secondary structures
There are three different types of secondary structures – also called primer dimers – that can form during a PCR assay:
- Hairpins: caused by intra-primer homology – when a region of three or more bases is complementary to another region within the same primer – or when a primer melting temperature is lower than the annealing temperature of the reaction.
- Self-dimers: formed when two same sense primers have complementary sequences – inter-primer homology – and anneal to each other.
- Cross-dimers: formed when forward and reverse primers anneal to each other when there is inter-primer homology.
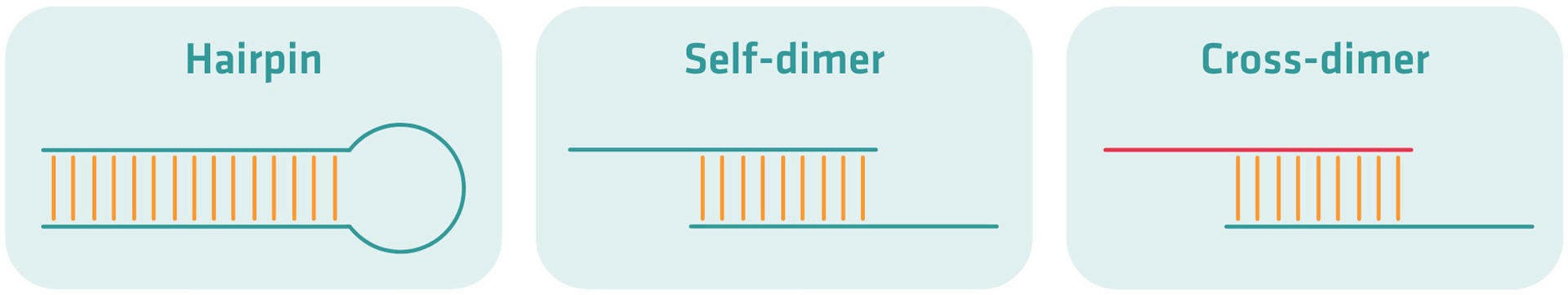
Your PCR product yield will be less if secondary structures form and remain stable above the annealing temperature of your reaction, as the primers bind to themselves or another primer instead of the template DNA. This is why your primer design tool should be able to check for, and warn you of stable secondary structures.
Mismatches and degenerated positions
Mismatches are primer bases that aren't complementary to the target sequence. They can be tolerated to a certain extent, and are sometimes even necessary; for example, when performing a multi-template PCR to amplify a set of similar target sequences from different bacteria with a single set of primers. Degenerate primers could help if mismatches negatively impact the performance of your PCR.
Degenerate primers have several different nucleotides in some of their positions. For example, instead of A you could have an equal concentration of A and T in a certain position. The codes for the different nucleotide combinations available for degenerate primers are as follows:
| IUPAC nucleotide code | Base |
|---|---|
| R | A or G |
| Y | C or T |
| S | G or C |
| W | A or T |
| K | G or T |
| M | A or C |
| B | C or G or T |
| D | A or G or T |
| H | A or C or T |
| V | A or C or G |
| N | Any base |
Conclusion
That's it, you're now a 'PCR Primer Pro'! If your PCR reaction still doesn't work as intended after designing your primers according to the guidelines above, check out our blog post on additional PCR tips and tricks, or the ideal PCR lab set-up to prevent contamination for more best practice guidelines.
Further reading:
Ask our expert. Leave a comment!
Write us if you have any questions regarding the blog article.